[Published online Journal of Computer Chemistry, Japan Vol.16, 110-111, by J-STAGE]
<Title:> Parameterization of Reactive Force Field for Iron Water System
<Author(s):> Qian CHEN, Jingxiang XU, Yusuke OOTANI, Nobuki OZAWA, Momoji KUBO
<Corresponding author E-Mill:> momoji(at)imr.tohoku.ac.jp
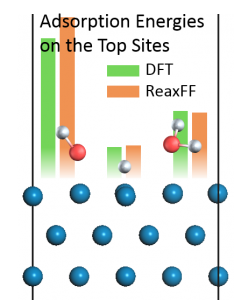
<Abstract:> Reactive force field parameters are re-optimized for simulating the stress corrosion cracking (SCC) of iron-based material in a supercritical water environment. The parameters for molecular dynamics (MD) simulation are determined by fitting the adsorption energies of H, OH, and H2O on an Fe(110) surface obtained by reactive force field to the density functional theory (DFT) calculations. The errors of adsorption energies for the most stable positions are less than 5%, and our parameters are in good agreement with the DFT calculations. The development of Fe/O/H parameters is expected to contribute to SCC simulation of iron-based materials in supercritical water environments.
<Keywords:> Molecular dynamics simulation, Reactive force field, DFT calculation, Iron-based material
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0041/_article/-char/ja/
<Title:> Parameterization of Reactive Force Field for Iron Water System
<Author(s):> Qian CHEN, Jingxiang XU, Yusuke OOTANI, Nobuki OZAWA, Momoji KUBO
<Corresponding author E-Mill:> momoji(at)imr.tohoku.ac.jp
<Abstract:> Reactive force field parameters are re-optimized for simulating the stress corrosion cracking (SCC) of iron-based material in a supercritical water environment. The parameters for molecular dynamics (MD) simulation are determined by fitting the adsorption energies of H, OH, and H2O on an Fe(110) surface obtained by reactive force field to the density functional theory (DFT) calculations. The errors of adsorption energies for the most stable positions are less than 5%, and our parameters are in good agreement with the DFT calculations. The development of Fe/O/H parameters is expected to contribute to SCC simulation of iron-based materials in supercritical water environments.
<Keywords:> Molecular dynamics simulation, Reactive force field, DFT calculation, Iron-based material
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0041/_article/-char/ja/