[Published online Journal of Computer Chemistry, Japan -International Edition Vol.7, -, by J-STAGE]
<Title:> Effect of meso-Substitution on the Selectivity of the Propene Reaction by Fe(IV)OCl Porphyrin: a Density Functional Theory Mechanistic Study
<Author(s):> Zhifeng MA, Masahiko HADA
<Corresponding author E-Mill:> hada(at)tmu.ac.jp
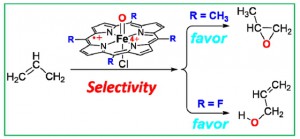
<Abstract:> We have performed density functional theory (DFT) calculations of C=C epoxidation and C-H hydroxylation of propene using a model of Fe(IV)OCl porphyrin cation radical complexes with fluorine and methyl groups as meso-substituents of the porphyrin ring. By gas-phase DFT calculations, it is found that fluorine substitution enhances the reactivity. According to detailed electronic feature analysis of the reactant complexes and transition states, electron-withdrawing groups at the meso-position stabilize the electron acceptor orbital of the complex more than the electron donation orbital of the substrate, leading to a decrease in the energy gap between these orbitals, and a lower energy barrier. More importantly, fluorine substitution for the pull effect makes hydroxylation favorable, whereas methyl substitution makes epoxidation preferable. The selective oxidation reactivity of Fe(IV)OCl porphyrin is largely ascribed to the effect of meso-substitution on the amount of electron transfer from propene to Fe(IV)OCl porphyrin. Additionally, we analyzed intersystem crossing between the quartet and sextet spin states using the potential energy surfaces (PESs), and the crossing seam between the quartet and sextet PESs occurs at around transition state TS1.
<Keywords:> Density functional theory, meso-substitution effect, Oxoiron(IV) porphyrin, Electronic structure, Molecular orbital
<URL:> https://www.jstage.jst.go.jp/article/jccjie/7/0/7_2020-0011/_html
<Title:> Effect of meso-Substitution on the Selectivity of the Propene Reaction by Fe(IV)OCl Porphyrin: a Density Functional Theory Mechanistic Study
<Author(s):> Zhifeng MA, Masahiko HADA
<Corresponding author E-Mill:> hada(at)tmu.ac.jp
<Abstract:> We have performed density functional theory (DFT) calculations of C=C epoxidation and C-H hydroxylation of propene using a model of Fe(IV)OCl porphyrin cation radical complexes with fluorine and methyl groups as meso-substituents of the porphyrin ring. By gas-phase DFT calculations, it is found that fluorine substitution enhances the reactivity. According to detailed electronic feature analysis of the reactant complexes and transition states, electron-withdrawing groups at the meso-position stabilize the electron acceptor orbital of the complex more than the electron donation orbital of the substrate, leading to a decrease in the energy gap between these orbitals, and a lower energy barrier. More importantly, fluorine substitution for the pull effect makes hydroxylation favorable, whereas methyl substitution makes epoxidation preferable. The selective oxidation reactivity of Fe(IV)OCl porphyrin is largely ascribed to the effect of meso-substitution on the amount of electron transfer from propene to Fe(IV)OCl porphyrin. Additionally, we analyzed intersystem crossing between the quartet and sextet spin states using the potential energy surfaces (PESs), and the crossing seam between the quartet and sextet PESs occurs at around transition state TS1.
<Keywords:> Density functional theory, meso-substitution effect, Oxoiron(IV) porphyrin, Electronic structure, Molecular orbital
<URL:> https://www.jstage.jst.go.jp/article/jccjie/7/0/7_2020-0011/_html