[Published online Journal of Computer Chemistry, Japan Vol.21, 52-54, by J-STAGE]
<Title:> 密度汎関数法におけるSCF計算条件の最適化による高速化
<Author(s):> 大田 栄二, 白幡 晃一, 石川 敦之
<Corresponding author E-Mill:> eiji(at)fujitsu.com
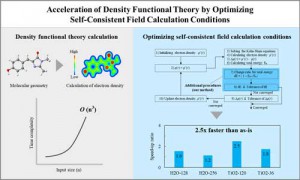
<Abstract:> Density functional theory (DFT) is a successful theory for calculating the electronic structure of atoms, molecules, and solids. However, in modern computing environments is difficult significantly improve the computational efficiency of DFT. Acceleration of DFT requires optimization of the computational algorithms. We demonstrate two acceleration methods by optimization of the computational algorithms that optimize parallel parameters for eigenvalue calculations and optimization of convergence conditions for the self-consistent field (SCF) calculation.
<Keywords:> Density functional theory, Self-consistent field, Eigenvalue calculation, Convergence conditions, Optimization
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/2/21_2022-0027/_article/-char/ja/
<Title:> 密度汎関数法におけるSCF計算条件の最適化による高速化
<Author(s):> 大田 栄二, 白幡 晃一, 石川 敦之
<Corresponding author E-Mill:> eiji(at)fujitsu.com
<Abstract:> Density functional theory (DFT) is a successful theory for calculating the electronic structure of atoms, molecules, and solids. However, in modern computing environments is difficult significantly improve the computational efficiency of DFT. Acceleration of DFT requires optimization of the computational algorithms. We demonstrate two acceleration methods by optimization of the computational algorithms that optimize parallel parameters for eigenvalue calculations and optimization of convergence conditions for the self-consistent field (SCF) calculation.
<Keywords:> Density functional theory, Self-consistent field, Eigenvalue calculation, Convergence conditions, Optimization
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/2/21_2022-0027/_article/-char/ja/