[Published online Journal of Computer Chemistry, Japan Vol.23, 27-29, by J-STAGE]
<Title:> アルキル鎖長が異なる有機半導体の電子物性差異に関する分子動力学および量子化学計算
<Author(s):> 岡田 智悠, 鈴木 朝香, 井上 悟, 長谷川 達生, 松井 弘之
<Corresponding author E-Mill:> t221488d(at)st.yamagata-u.ac.jp
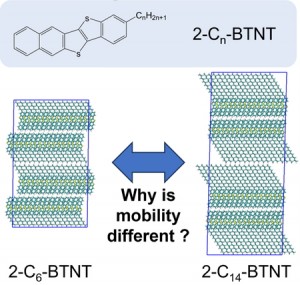
<Abstract:> The experimental mobilities of organic semiconductors, 2-Cn-BTNT (Figure 1), depend on the alkyl chain length n, while the mechanism is unclear. In this study, we elucidate the relationship between alkyl chain length and carrier mobility by classical force field (FF) and density functional theory (DFT) calculations. We analyzed the interactions between an alkyl chain and surrounding molecules and executed NVT molecular dynamics simulations at room temperature and ambient pressure by FF calculations. Transfer integrals were calculated using the DFT method with the coordinates after NVT simulations. As a result, alkyl chain length affects the lattice constant, thermal motion of the alkyl chain and transfer integrals and gives the difference of mobilities.
<Keywords:> キーワード Organic Semiconductors, Molecular Dynamics, Density Functional Theory, Transfer Integrals, Mobility
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2024-0005/_article/-char/ja/
<Title:> アルキル鎖長が異なる有機半導体の電子物性差異に関する分子動力学および量子化学計算
<Author(s):> 岡田 智悠, 鈴木 朝香, 井上 悟, 長谷川 達生, 松井 弘之
<Corresponding author E-Mill:> t221488d(at)st.yamagata-u.ac.jp
<Abstract:> The experimental mobilities of organic semiconductors, 2-Cn-BTNT (Figure 1), depend on the alkyl chain length n, while the mechanism is unclear. In this study, we elucidate the relationship between alkyl chain length and carrier mobility by classical force field (FF) and density functional theory (DFT) calculations. We analyzed the interactions between an alkyl chain and surrounding molecules and executed NVT molecular dynamics simulations at room temperature and ambient pressure by FF calculations. Transfer integrals were calculated using the DFT method with the coordinates after NVT simulations. As a result, alkyl chain length affects the lattice constant, thermal motion of the alkyl chain and transfer integrals and gives the difference of mobilities.
<Keywords:> キーワード Organic Semiconductors, Molecular Dynamics, Density Functional Theory, Transfer Integrals, Mobility
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/1/23_2024-0005/_article/-char/ja/