[Published online Journal of Computer Chemistry, Japan -International Edition Vol.10, -, by J-STAGE]
<Title:> Acceleration of Environmental Electrostatic Potential Using Cholesky Decomposition with Adaptive Metric (CDAM) for Fragment Molecular Orbital-based Molecular Dynamics (FMO-MD) Simulation
<Author(s):> Tatsuya NAKANO, Yoshio OKIYAMA, Katsunori SEGAWA, Yoshiro SAITO, Yuji MOCHIZUKI, Yuto KOMEIJI
<Corresponding author E-Mill:> y-komeiji(at)aist.go.jp
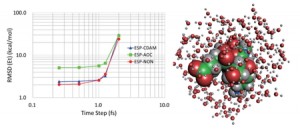
<Abstract:> The fragment molecular orbital (FMO) method is an efficient quantum chemical method suitable for calculating the electronic structures of large molecular systems. FMO can be accelerated by several approximations, an important one being the approximation to the environmental electrostatic potential (ESP) exerted on the fragment monomers or dimers. The environmental ESP is often approximated using the Mulliken atomic orbital charge (AOC) for proximal fragment dimers (ESP-AOC approximation) and the Mulliken point charge (PTC) for distant dimers (ESP-PTC approximation). Recently, another approximation method based on Cholesky decomposition with adaptive metric has been proposed for environmental ESP (ESP-CDAM, Okiyama et al., Bull. Chem. Soc. Jpn., 2021, 94, 91). In the current article, the energy gradient is derived under the ESP-CDAM approximation and implemented in FMO-based molecular dynamics (FMO-MD). Several test FMO-MD simulations are performed to compare the accuracy of the ESP-CDAM approximation with that of the conventional methods. The results show that ESP-CDAM is more accurate than ESP-AOC.
<Keywords:> Fragment molecular orbital method (FMO), Cholesky decomposition with adaptive metric (CDAM), Molecular dynamics, FMO-MD, ABINIT-MP
<URL:> https://www.jstage.jst.go.jp/article/jccjie/10/0/10_2023-0038/_html
<Title:> Acceleration of Environmental Electrostatic Potential Using Cholesky Decomposition with Adaptive Metric (CDAM) for Fragment Molecular Orbital-based Molecular Dynamics (FMO-MD) Simulation
<Author(s):> Tatsuya NAKANO, Yoshio OKIYAMA, Katsunori SEGAWA, Yoshiro SAITO, Yuji MOCHIZUKI, Yuto KOMEIJI
<Corresponding author E-Mill:> y-komeiji(at)aist.go.jp
<Abstract:> The fragment molecular orbital (FMO) method is an efficient quantum chemical method suitable for calculating the electronic structures of large molecular systems. FMO can be accelerated by several approximations, an important one being the approximation to the environmental electrostatic potential (ESP) exerted on the fragment monomers or dimers. The environmental ESP is often approximated using the Mulliken atomic orbital charge (AOC) for proximal fragment dimers (ESP-AOC approximation) and the Mulliken point charge (PTC) for distant dimers (ESP-PTC approximation). Recently, another approximation method based on Cholesky decomposition with adaptive metric has been proposed for environmental ESP (ESP-CDAM, Okiyama et al., Bull. Chem. Soc. Jpn., 2021, 94, 91). In the current article, the energy gradient is derived under the ESP-CDAM approximation and implemented in FMO-based molecular dynamics (FMO-MD). Several test FMO-MD simulations are performed to compare the accuracy of the ESP-CDAM approximation with that of the conventional methods. The results show that ESP-CDAM is more accurate than ESP-AOC.
<Keywords:> Fragment molecular orbital method (FMO), Cholesky decomposition with adaptive metric (CDAM), Molecular dynamics, FMO-MD, ABINIT-MP
<URL:> https://www.jstage.jst.go.jp/article/jccjie/10/0/10_2023-0038/_html