[Published online Journal of Computer Chemistry, Japan -International Edition1, 5-8, by J-STAGE]
<Title:> Conformational Analysis of Hexakis-N-Methylformamide Nickel(II) Complex on the Basis of Computational Group Theory and Density Functional Theory
<Author(s):> Hiroshi SAKIYAMA, Katsushi WAKI
<Corresponding author E-Mill:> saki(at)sci.kj.yamagata-u.ac.jp
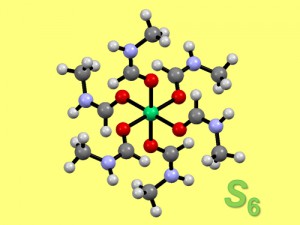
<Abstract:> Conformational analysis was conducted for a hexakis-N-methylformamide nickel(II) complex cation, [Ni- (NMF)6]2+ [hexakis(N-methylformamide-κO)nickel(II) dication], by the semi-empirical PM6 method and the Density Functional Theory (DFT) method, using 54 possible conformers obtained by the Computational Group Theory (CGT) method as initial structures. In the preliminary structural optimization by the PM6, all of the initial structures converted to only two conformers, pseudo–S6 and pseudo–D3 conformers. Subsequently, the structures of the two conformers were optimized by the DFT method, and the pseudo–S6 conformer was found to be more stable than the pseudo–D3 conformer by 1.8 ~ 2.2 kcal mol 1. From the energy difference, the pseudo–S6 conformer was found to be practically the only species at room temperature. This result is consistent with the crystal structure.
<Keywords:> Conformational analysis, Hexakis-N-methylformamide complex, Nickel(II) complex, Computational group theory (CGT) method, Density Functional Theory (DFT) method
<URL:> https://www.jstage.jst.go.jp/article/jccjie/1/0/1_2015-0047/_article
<Title:> Conformational Analysis of Hexakis-N-Methylformamide Nickel(II) Complex on the Basis of Computational Group Theory and Density Functional Theory
<Author(s):> Hiroshi SAKIYAMA, Katsushi WAKI
<Corresponding author E-Mill:> saki(at)sci.kj.yamagata-u.ac.jp
<Abstract:> Conformational analysis was conducted for a hexakis-N-methylformamide nickel(II) complex cation, [Ni- (NMF)6]2+ [hexakis(N-methylformamide-κO)nickel(II) dication], by the semi-empirical PM6 method and the Density Functional Theory (DFT) method, using 54 possible conformers obtained by the Computational Group Theory (CGT) method as initial structures. In the preliminary structural optimization by the PM6, all of the initial structures converted to only two conformers, pseudo–S6 and pseudo–D3 conformers. Subsequently, the structures of the two conformers were optimized by the DFT method, and the pseudo–S6 conformer was found to be more stable than the pseudo–D3 conformer by 1.8 ~ 2.2 kcal mol 1. From the energy difference, the pseudo–S6 conformer was found to be practically the only species at room temperature. This result is consistent with the crystal structure.
<Keywords:> Conformational analysis, Hexakis-N-methylformamide complex, Nickel(II) complex, Computational group theory (CGT) method, Density Functional Theory (DFT) method
<URL:> https://www.jstage.jst.go.jp/article/jccjie/1/0/1_2015-0047/_article