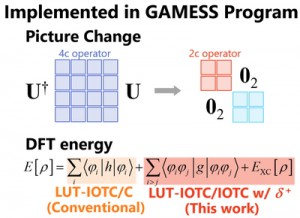
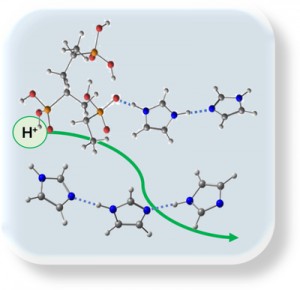
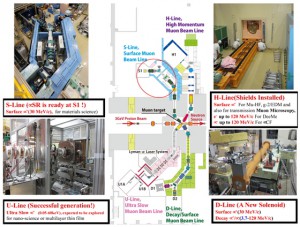
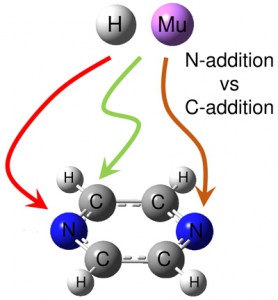
[Published online Journal of Computer Chemistry, Japan Vol.19, 146-148, by J-STAGE]
<Title:> β-LiAlSiO4結晶の温度変化シミュレーション
<Author(s):> 藤田 和希, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)mmm.muroran-it.ac.jp
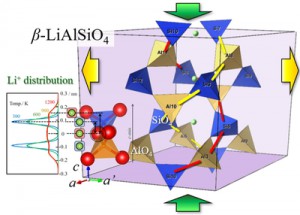
<Abstract:> Anisotropic low-thermal expansion of β-LiAlSiO4 crystal was reproduced by MD calculation applying a potential model of a spherically symmetric two-body interatomic interaction. From the correlation between the Si-O-Al angle and the Si-Al distance in the c-axis direction, the expansion of a-axis and the contract of c-axis brings the split of Si-O-Al angle distribution. At the same time, the stable position of Li+ ion sites also changed. This suggests the cation position also important in the mechanism of low-thermal expansion observed in silicate materials.
<Keywords:> Molecular dynamics, Beta-eucryptite, Thermal expansion, Bonding angle distribution
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0012/_article/-char/ja/
<Title:> β-LiAlSiO4結晶の温度変化シミュレーション
<Author(s):> 藤田 和希, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)mmm.muroran-it.ac.jp
<Abstract:> Anisotropic low-thermal expansion of β-LiAlSiO4 crystal was reproduced by MD calculation applying a potential model of a spherically symmetric two-body interatomic interaction. From the correlation between the Si-O-Al angle and the Si-Al distance in the c-axis direction, the expansion of a-axis and the contract of c-axis brings the split of Si-O-Al angle distribution. At the same time, the stable position of Li+ ion sites also changed. This suggests the cation position also important in the mechanism of low-thermal expansion observed in silicate materials.
<Keywords:> Molecular dynamics, Beta-eucryptite, Thermal expansion, Bonding angle distribution
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0012/_article/-char/ja/