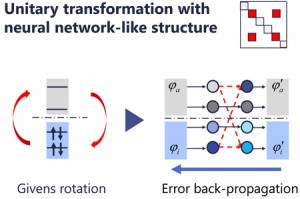
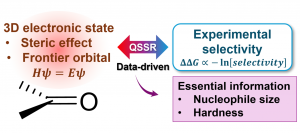
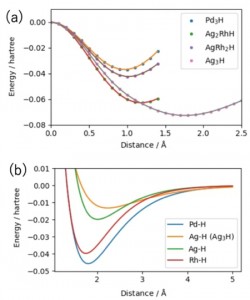
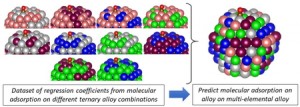
[Published online Journal of Computer Chemistry, Japan Vol.24, 58-67, by J-STAGE]
<Title:> 化学勾配の緩和過程に基づくゾウリムシの長距離後退遊泳行動モデリング
<Author(s):> 宇座 恩, 砂川 泰也, 國田 樹
<Corresponding author E-Mill:> kunita(at)ie.u-ryukyu.ac.jp
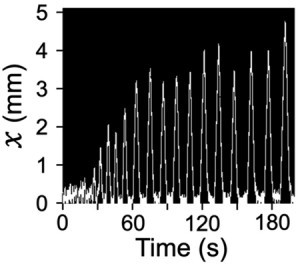
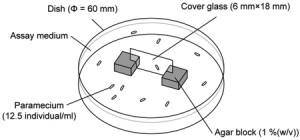
<Abstract:> 本研究は,ゾウリムシの空間適応行動を化学勾配の緩和過程に基づいて数理モデルで再現することを目的とした.ゾウリムシは,細長い空間内で障害物に衝突した際,短距離の後退遊泳を繰り返しながら徐々に後退距離を伸ばし,最終的に3-4 mmの長距離後退遊泳を発現する.この行動は細胞内外の化学勾配が膜電位を変化させ,繊毛運動の方向と頻度を制御することで生じる.そこで,膜電位動態と繊毛運動の関係を定式化した運動モデルを膜電位モデルに統合した数理モデルを構成した.数値シミュレーションにより,カルシウムイオンチャネルの遅い応答に加えて,後退遊泳を引き起こすカルシウム電流の閾値が動的に変化する仕組みを導入することで,短距離後退遊泳から1 mm程度までの長距離後退遊泳への移行過程の一部を再現できた.これにより,ゾウリムシは単に化学勾配に受動的に応答するだけでなく,膜電位と繊毛運動のダイナミクスを通じた内因的な制御メカニズムを備えた適応能を備えていることが示唆された.
<Keywords:> chemical gradients, membrane potential, Paramecium, ciliary reversal, Hodgkin Huxley model
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/2/24_2024-0035/_article/-char/ja/
<Title:> 化学勾配の緩和過程に基づくゾウリムシの長距離後退遊泳行動モデリング
<Author(s):> 宇座 恩, 砂川 泰也, 國田 樹
<Corresponding author E-Mill:> kunita(at)ie.u-ryukyu.ac.jp
<Abstract:> 本研究は,ゾウリムシの空間適応行動を化学勾配の緩和過程に基づいて数理モデルで再現することを目的とした.ゾウリムシは,細長い空間内で障害物に衝突した際,短距離の後退遊泳を繰り返しながら徐々に後退距離を伸ばし,最終的に3-4 mmの長距離後退遊泳を発現する.この行動は細胞内外の化学勾配が膜電位を変化させ,繊毛運動の方向と頻度を制御することで生じる.そこで,膜電位動態と繊毛運動の関係を定式化した運動モデルを膜電位モデルに統合した数理モデルを構成した.数値シミュレーションにより,カルシウムイオンチャネルの遅い応答に加えて,後退遊泳を引き起こすカルシウム電流の閾値が動的に変化する仕組みを導入することで,短距離後退遊泳から1 mm程度までの長距離後退遊泳への移行過程の一部を再現できた.これにより,ゾウリムシは単に化学勾配に受動的に応答するだけでなく,膜電位と繊毛運動のダイナミクスを通じた内因的な制御メカニズムを備えた適応能を備えていることが示唆された.
<Keywords:> chemical gradients, membrane potential, Paramecium, ciliary reversal, Hodgkin Huxley model
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/2/24_2024-0035/_article/-char/ja/