[Published online Journal of Computer Chemistry, Japan -International Edition Vol.6, -, by J-STAGE]
<Title:> QM- and ONIOM-Molecular Dynamics Studies of the SN2 Reaction. How Does the Rare Event Take Place?
<Author(s):> Toshiaki MATSUBARA
<Corresponding author E-Mill:> matsubara(at)kanagawa-u.ac.jp
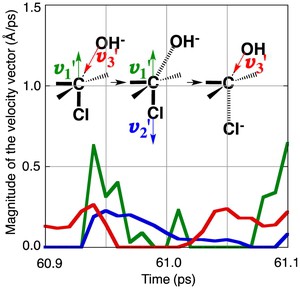
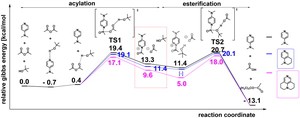
<Abstract:> A model SN2 reaction of 2-chlorobutane with OH– is examined from the dynamical point of view by means of the molecular dynamics (MD) methods. The reactions are simulated in both the gas phase and the water solvent by the QM- and the ONIOM-MD methods, respectively. The MD simulations show that a migration of the kinetic energy between the atoms generated by the fluctuation of the kinetic energy of the atoms triggers the reaction. When some requirements of the velocity vector with a certain direction of the specific atoms participating in the reaction are satisfied by the migration of the kinetic energy, the substrate is led to the transition state and then to the product in turn. The time required to pass the transition state becomes shorter in the water solvent compared to the case of the gas phase, which shows that the reaction proceeds more quickly in the water solvent. Another pattern of the requirement of the velocity vector with a certain direction of the specific atoms for the reaction, which makes the reaction rate faster, is also found at a higher temperature in the water solvent.
<Keywords:> Molecular dynamics simulation, SN2 reaction, Requirement for the reaction, Solvent effect
<URL:> https://www.jstage.jst.go.jp/article/jccjie/6/0/6_2019-0006/_html