[Published online Journal of Computer Chemistry, Japan Vol.18, 78-83, by J-STAGE]
<Title:> 第一原理計算を用いた硫化スズ電極のNaイオン電池性能評価と放電機構解明
<Author(s):> 小鷹 浩毅, 籾田 浩義, 喜多條 鮎子, 岡田 重人, 小口 多美夫
<Corresponding author E-Mill:> kotaka(at)esicb.kyoto-u.ac.jp
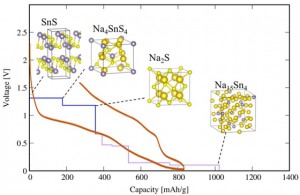
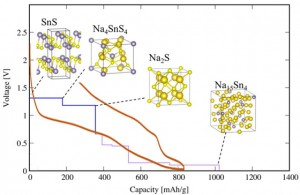
<Abstract:> スズ化合物は安価かつ大きな理論容量が期待されることから,Naイオン二次電池の負極材料候補として研究される物質の一つである.本研究では,Naイオン二次電池の負極材料となりうるコンバージョン系負極材料として硫化スズに着目し,その電池特性を第一原理計算を用いて調べた.負極材料とキャリアであるNaが反応した場合に生成すると予想されるNa-Sn-S系化合物を計算し,生成エネルギー解析に基づいて評価した3元系相図から充放電反応過程の生成物を明らかにした.計算から得た充放電反応式をもとに電圧容量曲線を作成し,Na / SnSハーフセル実験にて測定された充放電曲線との比較を行ったところ,実験結果をよく再現する結果を得た.さらに,充放電反応過程の生成物を特定するために,硫黄 K端のX線吸収スペクトルを計算し,実測結果と比較した.放電時にNa2S由来のスペクトル形状変化が現れ,それが充電時に再びSnS由来のスペクトル形状に戻ることを確認した.
<Keywords:> Na-ion battery, Electrode, First-principles calculation, Battery performance, X-ray absorption spectrum.
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/1/18_2018-0041/_article/-char/ja/