[Published online Journal of Computer Chemistry, Japan Vol.17, 127-129, by J-STAGE]
<Title:> 光受容タンパク質の機構解明に向けた分割統治型時間依存密度汎関数強束縛法の開発
<Author(s):> 河本 奈々, 吉川 武司, 小野 純一, 中井 浩巳
<Corresponding author E-Mill:> nakai(at)waseda.jp
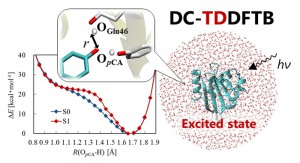
<Abstract:> The divide-and-conquer (DC) method was extended to time-dependent density functional tight-binding (TDDFTB) theory to enable excited-state calculations of large systems, as denoted by DC-TDDFTB. In this article, the implementation of TDDFTB and DC-TDDFTB methods into our in-house program, DC-DFTB-K, is explained. Dependence of CPU time on system-size for the DC-TDDFTB calculations indicates significant reduction of computational cost by adopting the DC method. Owing to the feature, excited-state calculations for whole photoactive yellow protein (PYP) surrounded by 4684 water molecules could be carried out in order to examine the hydrogen bond between p-coumaric acid and Glu46 in PYP.
<Keywords:> Divide-and-conquer method, Time-dependent density-functional tight-binding method, Photoactive yellow protein, Excited state, ??i??p??/i??-Coumaric acid.
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/3/17_2018-0032/_article/-char/ja/