[Published online Journal of Computer Chemistry, Japan Vol.17, 113-116, by J-STAGE]
<Title:> 分子動力学シミュレーションを用いたナトリウムホウ酸塩ガラスにおけるホウ酸異常現象の考察
<Author(s):> 山本 優也, 澤口 直哉, 佐々木 眞
<Corresponding author E-Mill:> 16096004(at)mmm.muroran-it.ac.jp
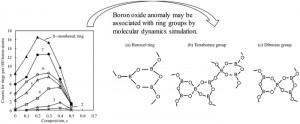
<Abstract:> We investigated the structural units of xNa2O-(1 – x) B2O3 glasses using molecular dynamics (MD) simulation with the interatomic potential provided by first-principles calculations. The results are consistent with experimental trends in interatomic distance, linear thermal expansion coefficient and BO4 units. The amount of 5- to 8-membered rings at x = 0.2 is larger than the other composition range. This suggests that the structural unit constructed by a few rings is related to suppression of thermal expansion.
<Keywords:> Molecular dynamics simulation, Borate glasses, Boron oxide anomaly, Oxide glasses
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/3/17_2018-0029/_article/-char/ja/