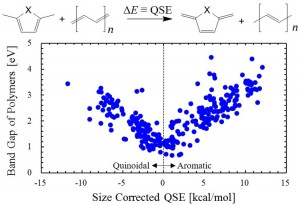
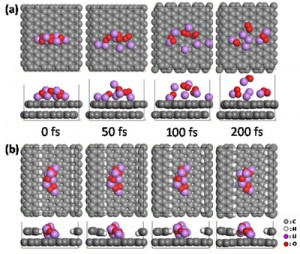
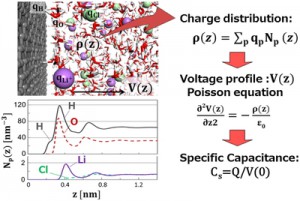
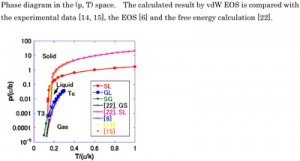
[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> フラグメント分子軌道(FMO)法を用いた散逸粒子動力学シミュレーションのための有効相互作用パラメータ算出の自動化フレームワーク
<Author(s):> 奥脇 弘次, 土居 英男, 望月 祐志
<Corresponding author E-Mill:> okuwaki(at)rikkyo.ac.jp
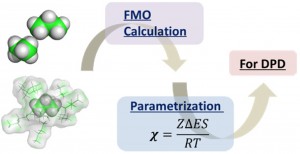
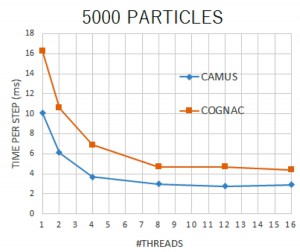
<Abstract:> 近年,用途に応じて目的の機能を有した材料開発の要求が高まっており,大規模な構造予測のためのシミュレーションが注目を浴びている.私達は,今回,散逸 粒子動力学(DPD: Dissipative Particle Dynamics)シミュレーションを例に,有効相互作用パラメータ(χパラメータと呼ばれる) をフラグメント分子軌道(FMO)法から算定するためのフレームワークとワークフローシステムを開発した.FMO計算は非経験的であるため,高分子系素材 に関わるほとんどの有機化合物に対して使うことが可能であり,χパラメータの算定に対して汎用性を持つと考えられる.この報告では,本システム (FCEWS: FMO-based Chi-parameter Evaluation Workflow System)の開発の目的,理論的な背景,ならびにソフトウェアについて解説する.
<Keywords:> Dissipative particle dynamics, DPD, fragment molecular orbital, FMO, chi-parameter
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2017-0048/_html/-char/ja/
<Title:> フラグメント分子軌道(FMO)法を用いた散逸粒子動力学シミュレーションのための有効相互作用パラメータ算出の自動化フレームワーク
<Author(s):> 奥脇 弘次, 土居 英男, 望月 祐志
<Corresponding author E-Mill:> okuwaki(at)rikkyo.ac.jp
<Abstract:> 近年,用途に応じて目的の機能を有した材料開発の要求が高まっており,大規模な構造予測のためのシミュレーションが注目を浴びている.私達は,今回,散逸 粒子動力学(DPD: Dissipative Particle Dynamics)シミュレーションを例に,有効相互作用パラメータ(χパラメータと呼ばれる) をフラグメント分子軌道(FMO)法から算定するためのフレームワークとワークフローシステムを開発した.FMO計算は非経験的であるため,高分子系素材 に関わるほとんどの有機化合物に対して使うことが可能であり,χパラメータの算定に対して汎用性を持つと考えられる.この報告では,本システム (FCEWS: FMO-based Chi-parameter Evaluation Workflow System)の開発の目的,理論的な背景,ならびにソフトウェアについて解説する.
<Keywords:> Dissipative particle dynamics, DPD, fragment molecular orbital, FMO, chi-parameter
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2017-0048/_html/-char/ja/