[Published online Journal of Computer Chemistry, Japan Vol.16, 77-79, by J-STAGE]
<Title:> ベンゼンを基本骨格に持つ正イオン内包ペプトイドの理論的計算
<Author(s):> 川田 修太郎, 袴田 真由, 望月 祐志
<Corresponding author E-Mill:> kawada(at)rikkyo.ac.jp
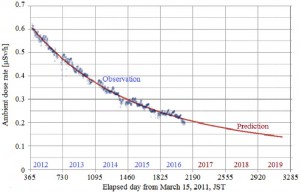
<Abstract:> ペプトイドは,側鎖がα-炭素原子上ではなく窒素原子上に位置するペプチド模倣物である.近年,主鎖にフェニル環を含む大環状のペプトイドが開発され,カ チオン捕捉の興味深い能力が示された.この論文では,この新しいペプトイドのフェニル環部によるカチオンの対捕捉に関する理論計算を報告する.こ の結果,カチオンに対するπ電子供与の重要性が示された.
<Keywords:> Peptoid, Cation capturing, Macrocycle, π-electron donation
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/3/16_2017-0014/_article/-char/ja/
<Title:> ベンゼンを基本骨格に持つ正イオン内包ペプトイドの理論的計算
<Author(s):> 川田 修太郎, 袴田 真由, 望月 祐志
<Corresponding author E-Mill:> kawada(at)rikkyo.ac.jp
<Abstract:> ペプトイドは,側鎖がα-炭素原子上ではなく窒素原子上に位置するペプチド模倣物である.近年,主鎖にフェニル環を含む大環状のペプトイドが開発され,カ チオン捕捉の興味深い能力が示された.この論文では,この新しいペプトイドのフェニル環部によるカチオンの対捕捉に関する理論計算を報告する.こ の結果,カチオンに対するπ電子供与の重要性が示された.
<Keywords:> Peptoid, Cation capturing, Macrocycle, π-electron donation
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/3/16_2017-0014/_article/-char/ja/
![]()