[Published online Journal of Computer Chemistry, Japan Vol.16, 28-31, by J-STAGE]
<Title:> 散逸粒子動力学におけるシリカ−脂質膜界面付近の水の取扱い
<Author(s):> 土居 英男, 奥脇 弘次, 望月 祐志, 小沢 拓
<Corresponding author E-Mill:> hideo-doi(at)rikkyo.ac.jp
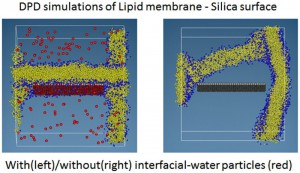
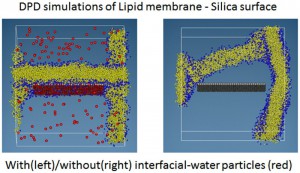
<Abstract:> 散逸粒子動力学(DPD: Dissipative Particle Dynamics)法を用いたシミュレーションは生体膜などの幅広い分野で用いられている.従来,このようなDPD シミュレーションでは,バルクの水と界面付近の水を同じように扱う場合がほとんどであった.しかし,こうしたやり方では系の性質を必ずしも忠実に再現して いるとはいえない.界面付近の水分子の性質とバルクの水分子の性質がかなり異なったものであるという実験的,あるいは理論的な研究がなされている からである.そこで,我々はこの問題を解決すべく,バルク水と界面水を区別して取り扱うアプローチを提案する.このアプローチを,脂質膜−シリカ −水の系のDPDシミュレーションに対して適用したところ,シリカに脂質膜が吸着する現象をうまくモデル化することができた.
<Keywords:> Dissipative particle dynamics, DPD, interface, water, lipid membrane
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/1/16_2017-0003/_article/-char/ja/
<Title:> 散逸粒子動力学におけるシリカ−脂質膜界面付近の水の取扱い
<Author(s):> 土居 英男, 奥脇 弘次, 望月 祐志, 小沢 拓
<Corresponding author E-Mill:> hideo-doi(at)rikkyo.ac.jp
<Abstract:> 散逸粒子動力学(DPD: Dissipative Particle Dynamics)法を用いたシミュレーションは生体膜などの幅広い分野で用いられている.従来,このようなDPD シミュレーションでは,バルクの水と界面付近の水を同じように扱う場合がほとんどであった.しかし,こうしたやり方では系の性質を必ずしも忠実に再現して いるとはいえない.界面付近の水分子の性質とバルクの水分子の性質がかなり異なったものであるという実験的,あるいは理論的な研究がなされている からである.そこで,我々はこの問題を解決すべく,バルク水と界面水を区別して取り扱うアプローチを提案する.このアプローチを,脂質膜−シリカ −水の系のDPDシミュレーションに対して適用したところ,シリカに脂質膜が吸着する現象をうまくモデル化することができた.
<Keywords:> Dissipative particle dynamics, DPD, interface, water, lipid membrane
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/1/16_2017-0003/_article/-char/ja/