[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> 多成分密度汎関数理論のための電子-核相関汎関数の開発
<Author(s):> 宇田川 太郎, 常田 貴夫, 立川 仁典
<Corresponding author E-Mill:> udagawa(at)gifu-u.ac.jp
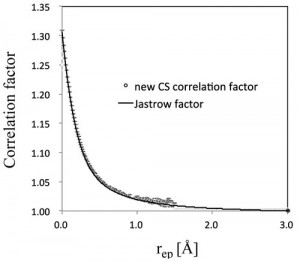
<Abstract:> 最も軽い水素の原子核の運動においては,量子効果が顕著に現れる. 原子核運動の量子効果を直接的に取り込んだ理論として多成分分子理論を開発してきた.また近年では,計算時間のかからない密度汎関数理論が分子理論の中心 理論となりつつある.我々は,多成分密度汎関数理論のための簡便な電子-核相関汎関数を開発した. この汎関数は,相関波動関数から簡単かつ正当な物理条件のみにもとづいて導出した汎用性の高い汎関数であり,パラメータを1つしか含まない.にもかかわら ず,水素原子を含む小分子の電子-プロトン相関エネルギーを化学的精度(1 2 kcal/mol程度の誤差)で再現する.
<Keywords:> Multicomponent density functional theory, electron-nucleus correlation functional, many-body effect, H/D isotope effect, correlations between different elementary particles
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2016-0018/_article/-char/ja/
<Title:> 多成分密度汎関数理論のための電子-核相関汎関数の開発
<Author(s):> 宇田川 太郎, 常田 貴夫, 立川 仁典
<Corresponding author E-Mill:> udagawa(at)gifu-u.ac.jp
<Abstract:> 最も軽い水素の原子核の運動においては,量子効果が顕著に現れる. 原子核運動の量子効果を直接的に取り込んだ理論として多成分分子理論を開発してきた.また近年では,計算時間のかからない密度汎関数理論が分子理論の中心 理論となりつつある.我々は,多成分密度汎関数理論のための簡便な電子-核相関汎関数を開発した. この汎関数は,相関波動関数から簡単かつ正当な物理条件のみにもとづいて導出した汎用性の高い汎関数であり,パラメータを1つしか含まない.にもかかわら ず,水素原子を含む小分子の電子-プロトン相関エネルギーを化学的精度(1 2 kcal/mol程度の誤差)で再現する.
<Keywords:> Multicomponent density functional theory, electron-nucleus correlation functional, many-body effect, H/D isotope effect, correlations between different elementary particles
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2016-0018/_article/-char/ja/