[Published online Journal of Computer Chemistry, Japan Vol.21, 99-102, by J-STAGE]
<Title:> 分子集合体の弾性体モデルにもとづく粗視化剛性行列の力学的解釈
<Author(s):> 北條 博彦, 中嶋 紘大, 岡村 彰太, 菊岡 龍太郎
<Corresponding author E-Mill:> houjou(at)iis.u-tokyo.ac.jp
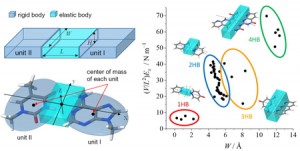
<Abstract:> We have been developing a method for coarse-graining the low-frequency vibration modes of molecular assemblies, which affords a numerical representation of the down-sized stiffness matrix. In this study, we present an analytical representation of the stiffness matrix based on the elastic-body modeling of molecular assemblies. Comparison between the numerical and analytical data allows the 13 parameters regarding the dimension and mechanical properties of the putative elastic body. The results for 57 molecular dimers with various hydrogen-bond multiplicity demonstrate that the obtained parameters were physically reasonable and well-reproduced the wavenumbers of normal-mode vibrations.
<Keywords:> Intermolecular stiffness, Normal-mode analysis, Elastic-body mechanics, Finite element method, Trans-scale simulation
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0006/_article/-char/ja/
<Title:> 分子集合体の弾性体モデルにもとづく粗視化剛性行列の力学的解釈
<Author(s):> 北條 博彦, 中嶋 紘大, 岡村 彰太, 菊岡 龍太郎
<Corresponding author E-Mill:> houjou(at)iis.u-tokyo.ac.jp
<Abstract:> We have been developing a method for coarse-graining the low-frequency vibration modes of molecular assemblies, which affords a numerical representation of the down-sized stiffness matrix. In this study, we present an analytical representation of the stiffness matrix based on the elastic-body modeling of molecular assemblies. Comparison between the numerical and analytical data allows the 13 parameters regarding the dimension and mechanical properties of the putative elastic body. The results for 57 molecular dimers with various hydrogen-bond multiplicity demonstrate that the obtained parameters were physically reasonable and well-reproduced the wavenumbers of normal-mode vibrations.
<Keywords:> Intermolecular stiffness, Normal-mode analysis, Elastic-body mechanics, Finite element method, Trans-scale simulation
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0006/_article/-char/ja/