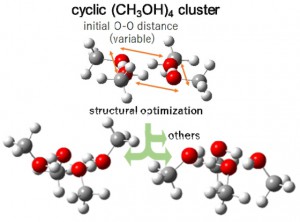
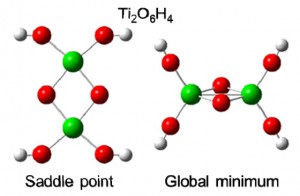
[Published online Journal of Computer Chemistry, Japan Vol.21, A19-A22, by J-STAGE]
<Title:> 千田顧問の遺したWinmostarとその思想
<Author(s):> 古賀 良太, 坂牧 隆司
<Corresponding author E-Mill:> rkoga(at)x-ability.jp
<Abstract:>
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/3/21_2022-0011/_article/-char/ja/
<Title:> 千田顧問の遺したWinmostarとその思想
<Author(s):> 古賀 良太, 坂牧 隆司
<Corresponding author E-Mill:> rkoga(at)x-ability.jp
<Abstract:>
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/3/21_2022-0011/_article/-char/ja/