[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> ケイ酸塩溶融体/ガラスのMD計算について- 無機物の分子シミュレーション-
<Author(s):> 河村 雄行
<Corresponding author E-Mill:> kawamura.k.ah(at)m.titech.ac.jp
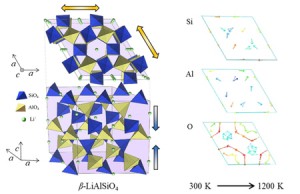
<Abstract:> 無機化合物のMD計算を念頭において,高分子様の構造を持つ無機凝集体について構造などの概要を示し,古典分子動力学法による2元系ケイ酸塩溶融体/ガラスの計算における構造緩和の問題を述べる.また無機凝集体のMD計算の困難さ,意義,および必要性について述べる.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2022-0012/_article/-char/ja/
<Title:> ケイ酸塩溶融体/ガラスのMD計算について- 無機物の分子シミュレーション-
<Author(s):> 河村 雄行
<Corresponding author E-Mill:> kawamura.k.ah(at)m.titech.ac.jp
<Abstract:> 無機化合物のMD計算を念頭において,高分子様の構造を持つ無機凝集体について構造などの概要を示し,古典分子動力学法による2元系ケイ酸塩溶融体/ガラスの計算における構造緩和の問題を述べる.また無機凝集体のMD計算の困難さ,意義,および必要性について述べる.
<Keywords:>
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2022-0012/_article/-char/ja/