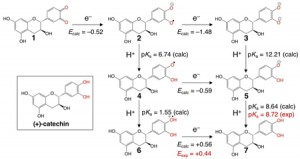
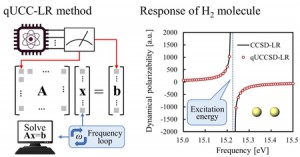
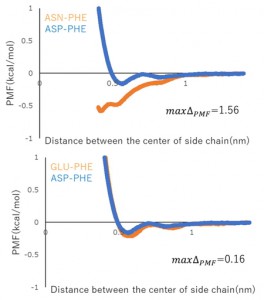
[Published online Journal of Computer Chemistry, Japan Vol.20, 129-131, by J-STAGE]
<Title:> 量子化学計算によるフェニルイソシアネート275 nm第一吸収帯の帰属
<Author(s):> 長沼 優人, 石橋 太祐, 奥山 克彦
<Corresponding author E-Mill:> okuyama.katsuhiko(at)nihon-u.ac.jp
<Abstract:> Abstract: Electronic spectra for the first 275-nm absorption system of phenyl isocyanate observed in a room temperature and in a jet were assigned with an assistance of quantum-chemistry calculation. All vibronic bands in a jet were diffuse with a width of 18 cm-1, indicating the lower limit of the life time was 204 fs. A suitable pair of a theoretical method and a basis set was selected among combinations of B3LYP, CAM-B3LYP, and MP2 and 18 kinds of basis sets on the basis of molecular-orbital energies and the rotational constant spectroscopically observed. B3LYP/cc-pVTZ and B3YP/aug-cc-pVDZ gave us good results. Time-dependent calculation was performed by two pairs. It was found out that the 275-nm absorption system was assigned as the first ππ* and third excited state and that twonπ* states with a very small oscillator strength were lying in the infrared and visible region. The fast relaxation process observed in a jet was due to an internal conversion to two nπ* states.
<Keywords:> Phenyl isocyanate, Quantum-chemistry calculation, Supersonic-jet spectroscopy, Symmetry assignyment, Absorption system
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/4/20_2021-0051/_article/-char/ja/
<Title:> 量子化学計算によるフェニルイソシアネート275 nm第一吸収帯の帰属
<Author(s):> 長沼 優人, 石橋 太祐, 奥山 克彦
<Corresponding author E-Mill:> okuyama.katsuhiko(at)nihon-u.ac.jp
<Abstract:> Abstract: Electronic spectra for the first 275-nm absorption system of phenyl isocyanate observed in a room temperature and in a jet were assigned with an assistance of quantum-chemistry calculation. All vibronic bands in a jet were diffuse with a width of 18 cm-1, indicating the lower limit of the life time was 204 fs. A suitable pair of a theoretical method and a basis set was selected among combinations of B3LYP, CAM-B3LYP, and MP2 and 18 kinds of basis sets on the basis of molecular-orbital energies and the rotational constant spectroscopically observed. B3LYP/cc-pVTZ and B3YP/aug-cc-pVDZ gave us good results. Time-dependent calculation was performed by two pairs. It was found out that the 275-nm absorption system was assigned as the first ππ* and third excited state and that twonπ* states with a very small oscillator strength were lying in the infrared and visible region. The fast relaxation process observed in a jet was due to an internal conversion to two nπ* states.
<Keywords:> Phenyl isocyanate, Quantum-chemistry calculation, Supersonic-jet spectroscopy, Symmetry assignyment, Absorption system
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/4/20_2021-0051/_article/-char/ja/