[Published online Journal of Computer Chemistry, Japan Vol.19, A12-A18, by J-STAGE]
<Title:> J-PARCミュオン施設 「J-PARC MUSE」
<Author(s):> 三宅 康博
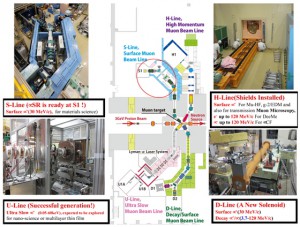
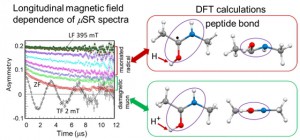
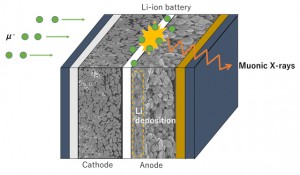
<Abstract:> ミュオン科学実験施設(MUSE)は,中性子, ハドロン, ニュートリノ施設とともに,2001年から2008年にかけて建設が許可されたJ-PARC計画の実験領域の一つである. MUSE施設は物質・生命科学実験施設(MLF)の中にあり,中性子とミュオンの両科学プログラムが統合されている. ミュオン製造用黒鉛ターゲットからパイオンやミュオンを効率的に取り出すための二次ミュオンラインは,Dライン, Uライン, Sライン, Hラインの4つのミュオンビームラインから構成されている.パルスミュオンビームの特徴を生かした10の実験エリア (D1, D2, U1A, U1B, S1, S2, S3, S4, H1, H2)において,様々なミュオン関連の実験を可能にしており,様々なシミュレーションコードを用いて,ビームラインの構成要素の設計を行っている.
<Keywords:> Keywords muon science facility (MUSE), J-PARC, Materials and Life Science Facility (MLF), pions, neutron and muon
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0016/_article/-char/ja/
<Title:> J-PARCミュオン施設 「J-PARC MUSE」
<Author(s):> 三宅 康博
<Abstract:> ミュオン科学実験施設(MUSE)は,中性子, ハドロン, ニュートリノ施設とともに,2001年から2008年にかけて建設が許可されたJ-PARC計画の実験領域の一つである. MUSE施設は物質・生命科学実験施設(MLF)の中にあり,中性子とミュオンの両科学プログラムが統合されている. ミュオン製造用黒鉛ターゲットからパイオンやミュオンを効率的に取り出すための二次ミュオンラインは,Dライン, Uライン, Sライン, Hラインの4つのミュオンビームラインから構成されている.パルスミュオンビームの特徴を生かした10の実験エリア (D1, D2, U1A, U1B, S1, S2, S3, S4, H1, H2)において,様々なミュオン関連の実験を可能にしており,様々なシミュレーションコードを用いて,ビームラインの構成要素の設計を行っている.
<Keywords:> Keywords muon science facility (MUSE), J-PARC, Materials and Life Science Facility (MLF), pions, neutron and muon
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0016/_article/-char/ja/