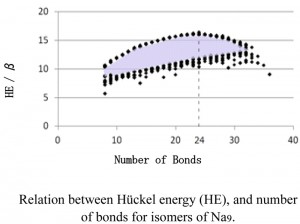
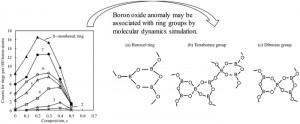
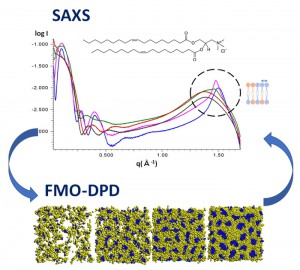
[Published online Journal of Computer Chemistry, Japan Vol.17, 120-121, by J-STAGE]
<Title:> ラジカル共重合の理論的意義付け
<Author(s):> 西久保 椋, 林 慶浩, 川内 進
<Corresponding author E-Mill:> skawauch(at)polymer.titech.ac.jp
<Abstract:> Radical copolymerization was theoretically investigated by using correlations between Q-e values and properties of monomers and terminal radical models. As a result, it was found that same e value used for monomers and radicals is reasonable. In addition, it was found that Q value is mainly related in reaction kinetics but not reaction thermodynamics.
<Keywords:> Polymer, Radical Copolymerization, Q-e Scheme
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/3/17_2018-0024/_article/-char/ja/
![]()