[Published online Journal of Computer Chemistry, Japan Vol.24, 77-79, by J-STAGE]
<Title:> Nonlocal Modulation of Antigen Antibody Interactions Underlying A107R-Mediated Affinity Gain in VHH D2-L29: MD Simulations with MM-PBSA Based Thermodynamic Analysis
<Author(s):> Rika MUNAKATA, Motoki INOUE, Takefumi YAMASHITA
<Corresponding author E-Mill:> yamashita.takefumi(at)hoshi.ac.jp
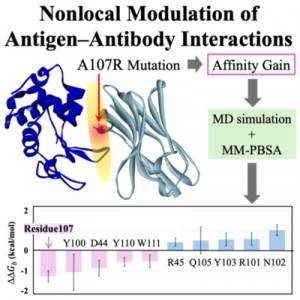
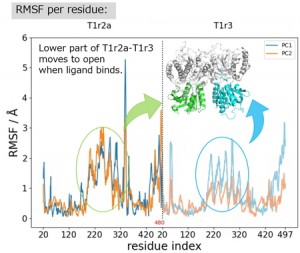
<Abstract:> Single-domain VHH antibodies offer attractive developability, but rational affinity improvement requires atomistic insight into how sequence changes reshape binding. We investigated the A107R substitution in VHH D2-L29 bound to hen egg-white lysozyme using long-timescale molecular dynamics simulations with MM-PBSA based thermodynamic analysis. Computed binding free energies recapitulated the experimentally observed increase in affinity. Per-residue decomposition of the change in binding free energy indicated that residue 107 (A107R) and Tyr100 had the largest favorable changes, whereas Asn102 had the largest unfavorable change. Together, the results indicate that molecularly detailed, interface-wide analysis of nonlocal interaction changes is essential for rationally engineering VHH variants with enhanced affinity.
<Keywords:> VHH, nanobodies, Molecular dynamics simulations, MM-PBSA, Affinity enhancement
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/3/24_2025-0015/_article/-char/ja/
<Title:> Nonlocal Modulation of Antigen Antibody Interactions Underlying A107R-Mediated Affinity Gain in VHH D2-L29: MD Simulations with MM-PBSA Based Thermodynamic Analysis
<Author(s):> Rika MUNAKATA, Motoki INOUE, Takefumi YAMASHITA
<Corresponding author E-Mill:> yamashita.takefumi(at)hoshi.ac.jp
<Abstract:> Single-domain VHH antibodies offer attractive developability, but rational affinity improvement requires atomistic insight into how sequence changes reshape binding. We investigated the A107R substitution in VHH D2-L29 bound to hen egg-white lysozyme using long-timescale molecular dynamics simulations with MM-PBSA based thermodynamic analysis. Computed binding free energies recapitulated the experimentally observed increase in affinity. Per-residue decomposition of the change in binding free energy indicated that residue 107 (A107R) and Tyr100 had the largest favorable changes, whereas Asn102 had the largest unfavorable change. Together, the results indicate that molecularly detailed, interface-wide analysis of nonlocal interaction changes is essential for rationally engineering VHH variants with enhanced affinity.
<Keywords:> VHH, nanobodies, Molecular dynamics simulations, MM-PBSA, Affinity enhancement
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/3/24_2025-0015/_article/-char/ja/