[Published online Journal of Computer Chemistry, Japan Vol.24, 17-19, by J-STAGE]
<Title:> アルキル鎖長が異なる有機半導体混合膜表面の分子動力学計算
<Author(s):> 鈴木 陸央, 宮田 稜, 井上 悟, 長谷川 達生, 松井 弘之
<Corresponding author E-Mill:> t231542d(at)st.yamagata-u.ac.jp
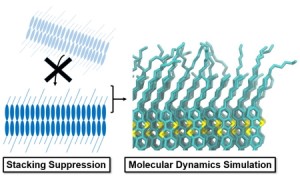
<Abstract:> A mixed solution of organic semiconductors Ph-BTBT-Cn with different alkyl chain length (n) forms a high-quality molecular bilayer by suppressing layer-by-layer stacking when the molar fraction of the longer chains (χL) is 0.1 0.6. In this study, we performed molecular dynamics simulations to investigate the dynamics of alkyl chains in the mixed bilayer. The order parameters and dihedral angles of the alkyl chains were analyzed as a function of χL. The results revealed that increasing χL enhances the ordering of the longer alkyl chains, thereby reducing their torsional motion. A stochastic model of the number of free surplus chains explains the molar fraction dependence of film morphology.
<Keywords:> キーワード Molecular dynamics, Organic semiconductor, Surface alkyl chain, Ordering, Interlayer frustration
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/1/24_2024-0038/_article/-char/ja/
<Title:> アルキル鎖長が異なる有機半導体混合膜表面の分子動力学計算
<Author(s):> 鈴木 陸央, 宮田 稜, 井上 悟, 長谷川 達生, 松井 弘之
<Corresponding author E-Mill:> t231542d(at)st.yamagata-u.ac.jp
<Abstract:> A mixed solution of organic semiconductors Ph-BTBT-Cn with different alkyl chain length (n) forms a high-quality molecular bilayer by suppressing layer-by-layer stacking when the molar fraction of the longer chains (χL) is 0.1 0.6. In this study, we performed molecular dynamics simulations to investigate the dynamics of alkyl chains in the mixed bilayer. The order parameters and dihedral angles of the alkyl chains were analyzed as a function of χL. The results revealed that increasing χL enhances the ordering of the longer alkyl chains, thereby reducing their torsional motion. A stochastic model of the number of free surplus chains explains the molar fraction dependence of film morphology.
<Keywords:> キーワード Molecular dynamics, Organic semiconductor, Surface alkyl chain, Ordering, Interlayer frustration
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/1/24_2024-0038/_article/-char/ja/