[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> 電子を描く(13) ― 混成軌道による分子の組立,σ結合とπ結合
<Author(s):> 時田 澄男
<Corresponding author E-Mill:> sumio.tokita(at)gmail.com
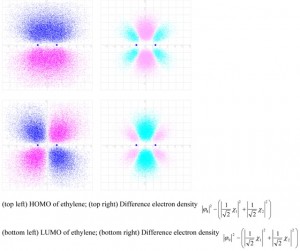
<Abstract:> 混成軌道を用いて,ルイスの電子対共有結合の概念にどのようにして同等性(等価性)や方向性を加味できるかを,簡単な分子を例にとって示す.σ電子とπ電子のちがい,π電子が核磁気共鳴スペクトルのケミカルシフトに及ぼす影響についても解説した.
<Keywords:> covalent bond, valence state, valence bond theory, molecular orbital method, hybrid orbital, resonance, difference electron density, NMR spectroscopy, ring current.
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2022-0021/_article/-char/ja/
<Title:> 電子を描く(13) ― 混成軌道による分子の組立,σ結合とπ結合
<Author(s):> 時田 澄男
<Corresponding author E-Mill:> sumio.tokita(at)gmail.com
<Abstract:> 混成軌道を用いて,ルイスの電子対共有結合の概念にどのようにして同等性(等価性)や方向性を加味できるかを,簡単な分子を例にとって示す.σ電子とπ電子のちがい,π電子が核磁気共鳴スペクトルのケミカルシフトに及ぼす影響についても解説した.
<Keywords:> covalent bond, valence state, valence bond theory, molecular orbital method, hybrid orbital, resonance, difference electron density, NMR spectroscopy, ring current.
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2022-0021/_article/-char/ja/