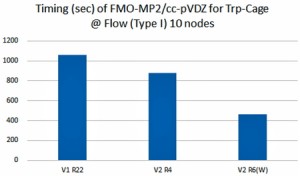
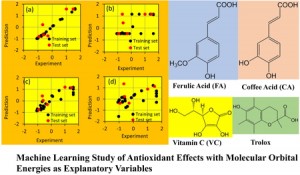
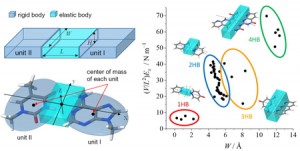
[Published online Journal of Computer Chemistry, Japan Vol.21, 134-136, by J-STAGE]
<Title:> シトクロムcの複合体形成過程の再現シミュレーション
<Author(s):> 下山 紘充, 重田 育照
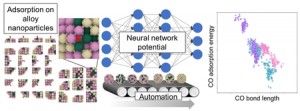
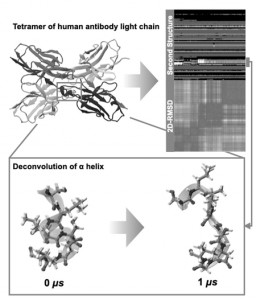
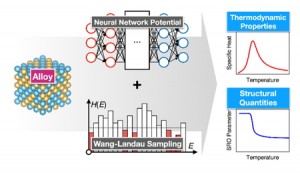
<Abstract:> A 3-dimensional domain swapping (3D-DS) phenomenon is a promising way to make a stable multimer because interactions in the multimer is about the same as that of a monomer; it would be useful to artificially design a functional protein’s multimer. In this study, molecular dynamics (MD) simulation of cytochrome c(cyt c) that is known to form a 3D-DS dimer (PDB ID: 3NBS) was performed to study factors that enhance 3D-DS structure sampling. Our results show a difficulty of 3D-DS structure sampling and a necessity of a method such as the generalized ensemble method. The results also show the importance of a loop-flexibility to sample 3D-DS structure; when the bond and torsion angle potential coefficients are less than 10% of secondary structure, 3D-DS structures become easy to sample.
<Keywords:> Cytochrome c, 3D-DS dimer, MD Simulation, Generalized ensemble method, Go-like model
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0014/_article/-char/ja/
<Title:> シトクロムcの複合体形成過程の再現シミュレーション
<Author(s):> 下山 紘充, 重田 育照
<Abstract:> A 3-dimensional domain swapping (3D-DS) phenomenon is a promising way to make a stable multimer because interactions in the multimer is about the same as that of a monomer; it would be useful to artificially design a functional protein’s multimer. In this study, molecular dynamics (MD) simulation of cytochrome c(cyt c) that is known to form a 3D-DS dimer (PDB ID: 3NBS) was performed to study factors that enhance 3D-DS structure sampling. Our results show a difficulty of 3D-DS structure sampling and a necessity of a method such as the generalized ensemble method. The results also show the importance of a loop-flexibility to sample 3D-DS structure; when the bond and torsion angle potential coefficients are less than 10% of secondary structure, 3D-DS structures become easy to sample.
<Keywords:> Cytochrome c, 3D-DS dimer, MD Simulation, Generalized ensemble method, Go-like model
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0014/_article/-char/ja/