月別: 2017年12月
分子動力学法による水系電解液電気二重層キャパシタの界面特性の解析 [Published online J. Comput. Chem. Jpn., 16, 112-115, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 112-115, by J-STAGE]
<Title:> 分子動力学法による水系電解液電気二重層キャパシタの界面特性の解析
<Author(s):> 矢野 振一郎, 古山 通久
<Corresponding author E-Mill:> koyama(at)ifrc.kyushu-u.ac.jp
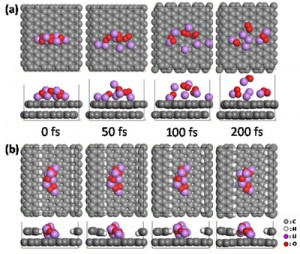
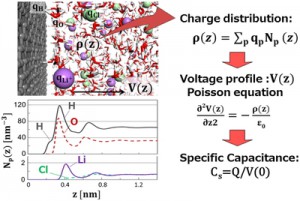
<Abstract:> Electric double layer capacitor (EDLC) is a storage device based on the interfacial interaction between electrode and electrolyte. We have conducted molecular dynamics simulations to study the atomistic origin of storage capacity. Electric double layer structures are studied by changing the electric field applied to the electrolyte. Charge density distribution in the electrolyte phase derived on the basis of molecular dynamics simulation is used to discuss the capacitance-voltage characteristics for different electrolyte species.
<Keywords:> キーワード:Electric double layer capacitor, Interfacial interaction, Molecular dynamics, Charge density distribution, Capacitance-voltage characteristics
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0033/_html/-char/ja/
<Title:> 分子動力学法による水系電解液電気二重層キャパシタの界面特性の解析
<Author(s):> 矢野 振一郎, 古山 通久
<Corresponding author E-Mill:> koyama(at)ifrc.kyushu-u.ac.jp
<Abstract:> Electric double layer capacitor (EDLC) is a storage device based on the interfacial interaction between electrode and electrolyte. We have conducted molecular dynamics simulations to study the atomistic origin of storage capacity. Electric double layer structures are studied by changing the electric field applied to the electrolyte. Charge density distribution in the electrolyte phase derived on the basis of molecular dynamics simulation is used to discuss the capacitance-voltage characteristics for different electrolyte species.
<Keywords:> キーワード:Electric double layer capacitor, Interfacial interaction, Molecular dynamics, Charge density distribution, Capacitance-voltage characteristics
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0033/_html/-char/ja/
ケクレ構造数え上げのアルゴリズムを用いたASC及びConjugated Circuitの計算プログラムの開発 [Published online in advanced , by J-STAGE]
[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> ケクレ構造数え上げのアルゴリズムを用いたASC及びConjugated Circuitの計算プログラムの開発
<Author(s):> 森川 , 野村 泰志, 溝口 則幸
<Corresponding author E-Mill:> dmorikawa(at)shinshu-u.ac.jp
<Abstract:> 成田らのケクレ構造の数え上げアルゴリズムを改良する事によって,各種共役分子のalgebraic structure count (ASC)を求める計算プログラムを開発した.さらに,この計算はケクレ構造の重ね合わせを利用する事から,全てのconjugated circuitの数え上げも可能となった.これによりASCとconjugated circuitを一度に求める事が可能となった.また,ASCを決定できない非交互共役系についても議論を行った.
<Keywords:> Algebraic structure count, Kekul structure, Conjugated circuit, Sachs graph, Resonance energy
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2017-0036/_html/-char/ja/
<Title:> ケクレ構造数え上げのアルゴリズムを用いたASC及びConjugated Circuitの計算プログラムの開発
<Author(s):> 森川 , 野村 泰志, 溝口 則幸
<Corresponding author E-Mill:> dmorikawa(at)shinshu-u.ac.jp
<Abstract:> 成田らのケクレ構造の数え上げアルゴリズムを改良する事によって,各種共役分子のalgebraic structure count (ASC)を求める計算プログラムを開発した.さらに,この計算はケクレ構造の重ね合わせを利用する事から,全てのconjugated circuitの数え上げも可能となった.これによりASCとconjugated circuitを一度に求める事が可能となった.また,ASCを決定できない非交互共役系についても議論を行った.
<Keywords:> Algebraic structure count, Kekul structure, Conjugated circuit, Sachs graph, Resonance energy
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2017-0036/_html/-char/ja/

Thermodynamics of Three-phase Equilibrium by Van Der Waals Equation of State [Published online J. Comput. Chem. Jpn. Int. Ed., 3, -, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan -International Edition Vol.3, -, by J-STAGE]
<Title:> Thermodynamics of Three-phase Equilibrium by Van Der Waals Equation of State
<Author(s):> Yosuke KATAOKA
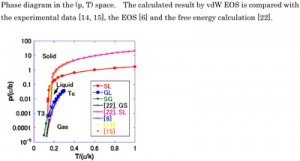
<Abstract:> The phase diagram for a three-phase equilibrium was derived using van der Waals (vdW) equation of state (EOS) with respect to pressure. Aside from the typical liquid-gas vdW EOS, a new solid-gas vdW EOS was introduced. The new vdW EOS had the same functional form as the original equation of state and only the van der Waals coefficients were different. The thermodynamic EOSs were integrated to obtain the internal energy and the integral constants were given explicitly. The calculated phase diagram was consistent with that for argon and the Lennard-Jones system.
<Keywords:> Three-phase equilibrium, Van der Waals equation of state, Argon, Lennard-Jones system, Thermodynamics
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0026/_html
<Title:> Thermodynamics of Three-phase Equilibrium by Van Der Waals Equation of State
<Author(s):> Yosuke KATAOKA
<Abstract:> The phase diagram for a three-phase equilibrium was derived using van der Waals (vdW) equation of state (EOS) with respect to pressure. Aside from the typical liquid-gas vdW EOS, a new solid-gas vdW EOS was introduced. The new vdW EOS had the same functional form as the original equation of state and only the van der Waals coefficients were different. The thermodynamic EOSs were integrated to obtain the internal energy and the integral constants were given explicitly. The calculated phase diagram was consistent with that for argon and the Lennard-Jones system.
<Keywords:> Three-phase equilibrium, Van der Waals equation of state, Argon, Lennard-Jones system, Thermodynamics
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0026/_html
Calculation of the Melting Entropy of Argon at Constant Volume Using Molecular Dynamics [Published online J. Comput. Chem. Jpn. Int. Ed., 3, -, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan -International Edition Vol.3, -, by J-STAGE]
<Title:> Calculation of the Melting Entropy of Argon at Constant Volume Using Molecular Dynamics
<Author(s):> Yosuke KATAOKA
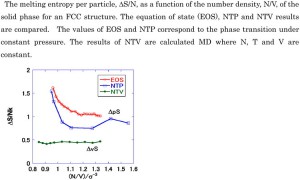
<Abstract:> The melting entropy values of argon at constant volume (ΔvS) as a function of density were calculated using molecular dynamics simulations. These calculations employed cells with N = 864 molecules. The resulting entropy of melting per particle, ΔvS/N, was found to be essentially constant (with some slight effect of the crystal form) and nearly equal to 0.5 k, where k is the Boltzmann constant. The melting entropy values at constant pressure (ΔpS) were also determined and it was observed that ΔpS > ΔvS. In addition, variations in the vibrational frequency in the liquid were compared with fluctuations in the solid near the melting point as a means of further investigating the melting entropy.
<Keywords:> Melting entropy at constant volume, Argon, Molecular dynamics, Fluctuation of vibrational frequency
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0021/_html
<Title:> Calculation of the Melting Entropy of Argon at Constant Volume Using Molecular Dynamics
<Author(s):> Yosuke KATAOKA
<Abstract:> The melting entropy values of argon at constant volume (ΔvS) as a function of density were calculated using molecular dynamics simulations. These calculations employed cells with N = 864 molecules. The resulting entropy of melting per particle, ΔvS/N, was found to be essentially constant (with some slight effect of the crystal form) and nearly equal to 0.5 k, where k is the Boltzmann constant. The melting entropy values at constant pressure (ΔpS) were also determined and it was observed that ΔpS > ΔvS. In addition, variations in the vibrational frequency in the liquid were compared with fluctuations in the solid near the melting point as a means of further investigating the melting entropy.
<Keywords:> Melting entropy at constant volume, Argon, Molecular dynamics, Fluctuation of vibrational frequency
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0021/_html
Parameterization of Reactive Force Field for Iron Water System [Published online J. Comput. Chem. Jpn., 16, 110-111, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 110-111, by J-STAGE]
<Title:> Parameterization of Reactive Force Field for Iron Water System
<Author(s):> Qian CHEN, Jingxiang XU, Yusuke OOTANI, Nobuki OZAWA, Momoji KUBO
<Corresponding author E-Mill:> momoji(at)imr.tohoku.ac.jp
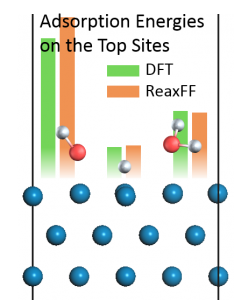
<Abstract:> Reactive force field parameters are re-optimized for simulating the stress corrosion cracking (SCC) of iron-based material in a supercritical water environment. The parameters for molecular dynamics (MD) simulation are determined by fitting the adsorption energies of H, OH, and H2O on an Fe(110) surface obtained by reactive force field to the density functional theory (DFT) calculations. The errors of adsorption energies for the most stable positions are less than 5%, and our parameters are in good agreement with the DFT calculations. The development of Fe/O/H parameters is expected to contribute to SCC simulation of iron-based materials in supercritical water environments.
<Keywords:> Molecular dynamics simulation, Reactive force field, DFT calculation, Iron-based material
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0041/_article/-char/ja/
<Title:> Parameterization of Reactive Force Field for Iron Water System
<Author(s):> Qian CHEN, Jingxiang XU, Yusuke OOTANI, Nobuki OZAWA, Momoji KUBO
<Corresponding author E-Mill:> momoji(at)imr.tohoku.ac.jp
<Abstract:> Reactive force field parameters are re-optimized for simulating the stress corrosion cracking (SCC) of iron-based material in a supercritical water environment. The parameters for molecular dynamics (MD) simulation are determined by fitting the adsorption energies of H, OH, and H2O on an Fe(110) surface obtained by reactive force field to the density functional theory (DFT) calculations. The errors of adsorption energies for the most stable positions are less than 5%, and our parameters are in good agreement with the DFT calculations. The development of Fe/O/H parameters is expected to contribute to SCC simulation of iron-based materials in supercritical water environments.
<Keywords:> Molecular dynamics simulation, Reactive force field, DFT calculation, Iron-based material
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/4/16_2017-0041/_article/-char/ja/