[Published online Journal of Computer Chemistry, Japan Vol.18, 152-155, by J-STAGE]
<Title:> インフォマティクス手法を活用した結合エネルギー密度解析の開発
<Author(s):> 中村 海里, 清野 淳司, 中井 浩巳
<Corresponding author E-Mill:> nakai(at)waseda.jp
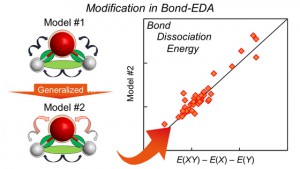
<Abstract:> The bond energy density analysis (bond-EDA), which is one of the two-body energy decomposition schemes to obtain bond energies, was extended by revisiting the formula and the constraint conditions, and using the informatics technique. Numerical assessments of the present schemes were performed for 44 chemical bonds in 33 small molecules with covalent and ionic bonds involving second and third row atoms. The results show that the present scheme qualitatively reproduces the bond dissociation energies obtained by the direct evaluation scheme in quantum chemical calculations.
<Keywords:> Bond energy density analysis, Chemical bond, Bond energy, Bond dissociation energy, Quantum chemical calculation
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/3/18_2019-0026/_article/-char/ja/