[Published online Journal of Computer Chemistry, Japan Vol.18, 166-168, by J-STAGE]
<Title:> Pd触媒を用いたアリルウレタンの分子内ヒドロアミノ化反応機構の理論的解明
<Author(s):> 河田 悠太, 林 慶浩, 高田 十志和, 川内 進
<Corresponding author E-Mill:> kawauchi.s.aa(at)m.titech.ac.jp
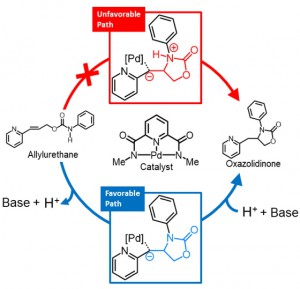
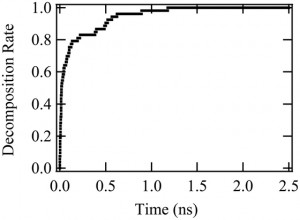
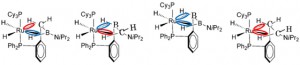
<Abstract:> Intramolecular hydroamination of allylurethane using the Pd-centered macrocycle catalyst was reported as an efficient continuous modification of oligo-functional allylurethanes. To clarify the mechanism of the hydroamination, quantum chemical calculation was performed for the model reaction. As a result, the cyclization in the hydroamination occurs via the initial deprotonation from the urethane N-H. In addition, the deprotonation was suggested to be a driving force for the transfer of the macrocyclic catalyst.
<Keywords:> 分子内ヒドロアミノ化, 量子化学計算, 反応機構, ロタキサン
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/3/18_2019-0014/_article/-char/ja/