[Published online Journal of Computer Chemistry, Japan Vol.20, 1-9, by J-STAGE]
<Title:> 化合物のAmes予測におけるGraph Convolutional Networkの特徴評価
<Author(s):> 半田 千彰, 小沢 知永, 福澤 薫, 米持 悦生
<Corresponding author E-Mill:> chiaki_handa(at)pharm.kissei.co.jp
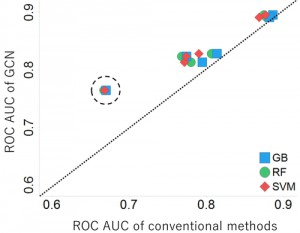
<Abstract:> 医薬品候補物質の潜在的な発がん性早期警戒システムであるAmes試験のin silico予測は,創薬研究において重要な予測項目の一つである.in silico予測の一手法である機械学習による予測では,Applicability Domain (AD)という機械学習モデルが本来の性能を発揮できるデータ領域を定義する研究がある.創薬研究においては,学習データと構造類似性が低い医薬品候補化合物の予測を行う場合があり,そのような化合物はAD領域外になる可能性が高く予測精度が低下する傾向がある.本研究では,Ames試験の機械予測モデルを作成し,テストデータとしてAD領域内/外となる確率が高い化合物群をそれぞれ用意して,複数の機械学習手法の予測性能を評価した.人工知能技術の発展により,創薬分野でも注目を集めているGraph Convolutional Network (GCN)と既存の機械学習手法の予測性能を比較した結果,AD領域外となる可能性が高い化合物群の予測性能において,GCNは既存手法より優れていた.
<Keywords:> Keywords Graph Convolutional Network, Machine learning, Ames test, Applicability Domain, Structural similarity
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/1/20_2020-0015/_article/-char/ja/
<Title:> 化合物のAmes予測におけるGraph Convolutional Networkの特徴評価
<Author(s):> 半田 千彰, 小沢 知永, 福澤 薫, 米持 悦生
<Corresponding author E-Mill:> chiaki_handa(at)pharm.kissei.co.jp
<Abstract:> 医薬品候補物質の潜在的な発がん性早期警戒システムであるAmes試験のin silico予測は,創薬研究において重要な予測項目の一つである.in silico予測の一手法である機械学習による予測では,Applicability Domain (AD)という機械学習モデルが本来の性能を発揮できるデータ領域を定義する研究がある.創薬研究においては,学習データと構造類似性が低い医薬品候補化合物の予測を行う場合があり,そのような化合物はAD領域外になる可能性が高く予測精度が低下する傾向がある.本研究では,Ames試験の機械予測モデルを作成し,テストデータとしてAD領域内/外となる確率が高い化合物群をそれぞれ用意して,複数の機械学習手法の予測性能を評価した.人工知能技術の発展により,創薬分野でも注目を集めているGraph Convolutional Network (GCN)と既存の機械学習手法の予測性能を比較した結果,AD領域外となる可能性が高い化合物群の予測性能において,GCNは既存手法より優れていた.
<Keywords:> Keywords Graph Convolutional Network, Machine learning, Ames test, Applicability Domain, Structural similarity
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/1/20_2020-0015/_article/-char/ja/