[Published online Journal of Computer Chemistry, Japan Vol.20, 106-111, by J-STAGE]
<Title:> 機械学習QSARの整数計画法に基づく逆解析法
<Author(s):> 仁 永持, 見深 朱, Naveed Ahmed AZAM, 和也 原口, 亮 趙, 達也 阿久津
<Corresponding author E-Mill:> nag(at)amp.i.kyoto-u.ac.jp
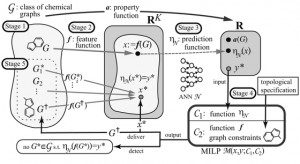
<Abstract:> We have developed a novel framework for inferring a chemical graph. Given an ANN that maps a feature vector x = f ( G ) of a chemical graph G to a predicted value η ( x ) of a chemical property to G, we formulate an integer program that simulates the functions f and η. Given a target value y*, we can infer a chemical graph G such that η ( f ( G ) ) = y * by solving the integer program.
<Keywords:> Machine learning, Integer programming, Chemical graphs, QSAR/QSPR, Molecular design
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0030/_article/-char/ja/
<Title:> 機械学習QSARの整数計画法に基づく逆解析法
<Author(s):> 仁 永持, 見深 朱, Naveed Ahmed AZAM, 和也 原口, 亮 趙, 達也 阿久津
<Corresponding author E-Mill:> nag(at)amp.i.kyoto-u.ac.jp
<Abstract:> We have developed a novel framework for inferring a chemical graph. Given an ANN that maps a feature vector x = f ( G ) of a chemical graph G to a predicted value η ( x ) of a chemical property to G, we formulate an integer program that simulates the functions f and η. Given a target value y*, we can infer a chemical graph G such that η ( f ( G ) ) = y * by solving the integer program.
<Keywords:> Machine learning, Integer programming, Chemical graphs, QSAR/QSPR, Molecular design
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0030/_article/-char/ja/