[Published online Journal of Computer Chemistry, Japan Vol.21, 80-81, by J-STAGE]
<Title:> 有機分子触媒を用いたアミノ酸ラセミ化反応の理論的研究
<Author(s):> 渡辺 七都稀, 庄司 光男, 堀 優太, 重田 育照
<Corresponding author E-Mill:> watanabe.natsuki.sm(at)alumni.tsukuba.ac.jp
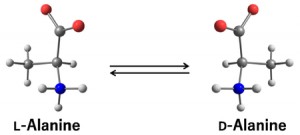
<Abstract:> Racemization of amino acid catalyzed by aldehyde and carboxylic acid has been proposed and widely used. Herein, we investigated the detailed racemization mechanism of alanine catalyzed by salicylaldehyde and acetic acid. Quantum chemistry calculations based on the density functional theory demonstrated that the dehydration reaction is the rate-determining step, and this dehydration step is enhanced by surrounding water molecules. We also discussed the dependence of the reaction rates on the type of amino acid.
<Keywords:> Racemization reaction, Organocatalytic reaction, Amino acid, Alanine, Density functional theory
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0004/_article/-char/ja/
<Title:> 有機分子触媒を用いたアミノ酸ラセミ化反応の理論的研究
<Author(s):> 渡辺 七都稀, 庄司 光男, 堀 優太, 重田 育照
<Corresponding author E-Mill:> watanabe.natsuki.sm(at)alumni.tsukuba.ac.jp
<Abstract:> Racemization of amino acid catalyzed by aldehyde and carboxylic acid has been proposed and widely used. Herein, we investigated the detailed racemization mechanism of alanine catalyzed by salicylaldehyde and acetic acid. Quantum chemistry calculations based on the density functional theory demonstrated that the dehydration reaction is the rate-determining step, and this dehydration step is enhanced by surrounding water molecules. We also discussed the dependence of the reaction rates on the type of amino acid.
<Keywords:> Racemization reaction, Organocatalytic reaction, Amino acid, Alanine, Density functional theory
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2023-0004/_article/-char/ja/