[Published online Journal of Computer Chemistry, Japan Vol.21, 106-110, by J-STAGE]
<Title:> FMOプログラムABINIT-MPの整備状況2022
<Author(s):> 望月 祐志, 中野 達也, 坂倉 耕太, 渡邊 啓正, 佐藤 伸哉, 奥脇 弘次, 秋澤 和輝, 土居 英男, 大島 聡史, 片桐 孝洋
<Corresponding author E-Mill:> fullmoon(at)rikkyo.ac.jp
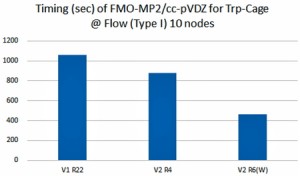
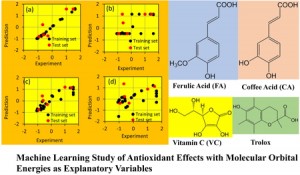
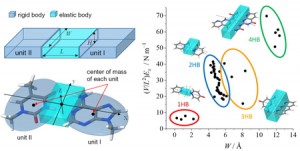
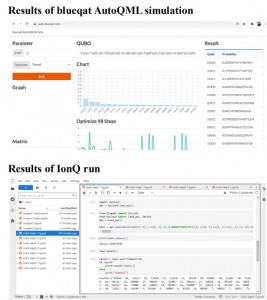
<Abstract:> We have been developing the ABINIT-MP program for fragment molecular orbital (FMO) calculations over 20 years. Several improvements for accelerated processing were made after the release of Open Version 2 Revision 4 at September 2021. Functionalities were enhanced as well. In this short report, we summarize such developments toward the next release of Revision 8.
<Keywords:> Fragment molecular orbital, FMO, ABINIT-MP, Supercomputer, A64FX, SX-Aurora TSUBASA
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2022-0037/_article/-char/ja/
<Title:> FMOプログラムABINIT-MPの整備状況2022
<Author(s):> 望月 祐志, 中野 達也, 坂倉 耕太, 渡邊 啓正, 佐藤 伸哉, 奥脇 弘次, 秋澤 和輝, 土居 英男, 大島 聡史, 片桐 孝洋
<Corresponding author E-Mill:> fullmoon(at)rikkyo.ac.jp
<Abstract:> We have been developing the ABINIT-MP program for fragment molecular orbital (FMO) calculations over 20 years. Several improvements for accelerated processing were made after the release of Open Version 2 Revision 4 at September 2021. Functionalities were enhanced as well. In this short report, we summarize such developments toward the next release of Revision 8.
<Keywords:> Fragment molecular orbital, FMO, ABINIT-MP, Supercomputer, A64FX, SX-Aurora TSUBASA
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/4/21_2022-0037/_article/-char/ja/