[Published online Journal of Computer Chemistry, Japan Vol.23, 59-61, by J-STAGE]
<Title:> Wulffの定理と第一原理計算を用いた金属クラスターの構造予測
<Author(s):> 大西 未優, 大野 彰太, 中田 彩子, 中井 浩巳
<Corresponding author E-Mill:> nakai(at)waseda.jp
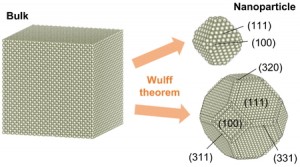
<Abstract:> Metal nanoparticles are useful as catalysts having specific reactivity owing to highly reactive site and strong size dependency. Structural information of metal nanoparticles is essential for interpretation and prediction of their reactivity. Wulff theorem predicts the equilibrium structures of crystals by using the surface energies of plane indices such as (111), (110), and (100). In this study, we evaluated the surface energies of well-defined Rh surfaces by the first principles calculations, followed by systematically constructing various sizes of Rh nanoparticles based on the Wulff theorem. For small nanoparticles with radii of 2 nm or less, only the (111) and (100) planes were present. On the other hand, high index surfaces appeared at large nanoparticles, of which the radii were more than 2.5 nm.
<Keywords:> Metal nanoparticle, Wulff construction, First principles calculation, Surface energy, Plane index
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/3/23_2024-0023/_article/-char/ja/
<Title:> Wulffの定理と第一原理計算を用いた金属クラスターの構造予測
<Author(s):> 大西 未優, 大野 彰太, 中田 彩子, 中井 浩巳
<Corresponding author E-Mill:> nakai(at)waseda.jp
<Abstract:> Metal nanoparticles are useful as catalysts having specific reactivity owing to highly reactive site and strong size dependency. Structural information of metal nanoparticles is essential for interpretation and prediction of their reactivity. Wulff theorem predicts the equilibrium structures of crystals by using the surface energies of plane indices such as (111), (110), and (100). In this study, we evaluated the surface energies of well-defined Rh surfaces by the first principles calculations, followed by systematically constructing various sizes of Rh nanoparticles based on the Wulff theorem. For small nanoparticles with radii of 2 nm or less, only the (111) and (100) planes were present. On the other hand, high index surfaces appeared at large nanoparticles, of which the radii were more than 2.5 nm.
<Keywords:> Metal nanoparticle, Wulff construction, First principles calculation, Surface energy, Plane index
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/3/23_2024-0023/_article/-char/ja/