[Published online Journal of Computer Chemistry, Japan Vol.23, 71-74, by J-STAGE]
<Title:> 分子動力学計算を用いたNa2O-K2O-SiO2系ガラスにおける混合アルカリ効果の再現
<Author(s):> 吉本 直樹, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)muroran-it.ac.jp
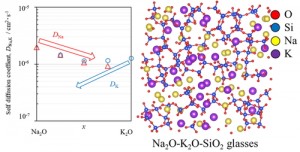
<Abstract:> Molecular dynamics simulation of y{(1-x)Na2O-xK2O}-(1-y)SiO2 glasses used an improved interatomic potential was performed to investigate the mixed alkali effect. The relation of self-diffusion coefficient of potassium and of sodium was improved, but the trend with x of the self-diffusion coefficient of potassium has become worse than the previous work.
<Keywords:> Keywords Molecular dynamics, Silicate glass, Mixed-Alkali Effect
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/3/23_2024-0025/_article/-char/ja/
<Title:> 分子動力学計算を用いたNa2O-K2O-SiO2系ガラスにおける混合アルカリ効果の再現
<Author(s):> 吉本 直樹, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)muroran-it.ac.jp
<Abstract:> Molecular dynamics simulation of y{(1-x)Na2O-xK2O}-(1-y)SiO2 glasses used an improved interatomic potential was performed to investigate the mixed alkali effect. The relation of self-diffusion coefficient of potassium and of sodium was improved, but the trend with x of the self-diffusion coefficient of potassium has become worse than the previous work.
<Keywords:> Keywords Molecular dynamics, Silicate glass, Mixed-Alkali Effect
<URL:> https://www.jstage.jst.go.jp/article/jccj/23/3/23_2024-0025/_article/-char/ja/