[Published online Journal of Computer Chemistry, Japan Vol.24, 5-9, by J-STAGE]
<Title:> 光2重イオン化されたOCS分子のダイナミクスと隠される反応経路
<Author(s):> 神原 龍冬, 堤 拓朗, 古屋 謙治, 武次 徹也
<Corresponding author E-Mill:> take(at)sci.hokudai.ac.jp
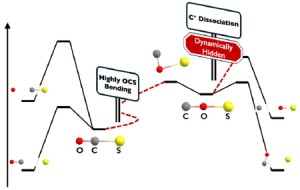
<Abstract:> The global reaction route mapping (GRRM) strategy enables an exhaustive search of static chemical reaction pathways, providing deep insight into chemical reactions. On the other hand, ab initio molecular dynamics (AIMD) simulations explicitly account for the momenta of atoms, revealing the realistic dynamical motion of molecules. In this study, we constructed the reaction path network for the ground-state OCS2+ species, which has attracted significant interest from spectroscopists. Recently, a new dissociation mechanism of S+via the COS2+ isomer has been proposed. By referencing the results of AIMD simulations and focusing on the isomerization process, we emphasize the crucial role of dynamical effects and the concept of dynamically hidden reaction paths.
<Keywords:> Keywords Reaction path network, Ab initio molecular dynamics, Dynamically hidden reaction path
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/1/24_2024-0044/_article/-char/ja/
<Title:> 光2重イオン化されたOCS分子のダイナミクスと隠される反応経路
<Author(s):> 神原 龍冬, 堤 拓朗, 古屋 謙治, 武次 徹也
<Corresponding author E-Mill:> take(at)sci.hokudai.ac.jp
<Abstract:> The global reaction route mapping (GRRM) strategy enables an exhaustive search of static chemical reaction pathways, providing deep insight into chemical reactions. On the other hand, ab initio molecular dynamics (AIMD) simulations explicitly account for the momenta of atoms, revealing the realistic dynamical motion of molecules. In this study, we constructed the reaction path network for the ground-state OCS2+ species, which has attracted significant interest from spectroscopists. Recently, a new dissociation mechanism of S+via the COS2+ isomer has been proposed. By referencing the results of AIMD simulations and focusing on the isomerization process, we emphasize the crucial role of dynamical effects and the concept of dynamically hidden reaction paths.
<Keywords:> Keywords Reaction path network, Ab initio molecular dynamics, Dynamically hidden reaction path
<URL:> https://www.jstage.jst.go.jp/article/jccj/24/1/24_2024-0044/_article/-char/ja/