[Published online Journal of Computer Chemistry, Japan Vol.20, 100-102, by J-STAGE]
<Title:> 3次元畳み込みニューラルネットワークの転移学習を用いたブロック共重合体の応力ひずみ曲線予測
<Author(s):> 川口 裕靖, 伊藤 真利子, 青柳 岳司, 大西 立顕
<Corresponding author E-Mill:> hironobukawaguchi(at)rikkyo.ac.jp
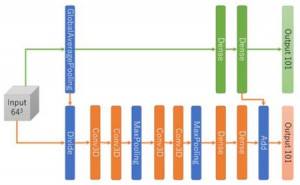
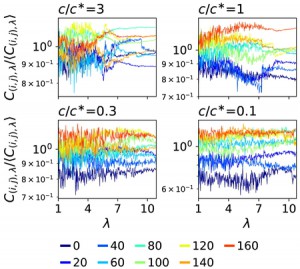
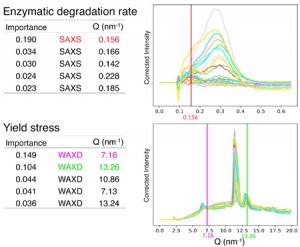
<Abstract:> The simulation to obtain stress-strain curves of block copolymers requires large computational resources. As an alternative to simulation, a method for high-throughput prediction of stress-strain curves using a three-dimensional convolutional neural network has been reported. In this study, we incorporated shortcut coupling into the neural network and performed pre-training and transfer learning in a step-by-step manner to successfully predict the stress-strain curve with high accuracy while reducing the training cost.
<Keywords:> Block copolymers, Microphase separation, Stress-strain curve, Deep learning, 3D convolutional neural network
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0037/_article/-char/ja/
<Title:> 3次元畳み込みニューラルネットワークの転移学習を用いたブロック共重合体の応力ひずみ曲線予測
<Author(s):> 川口 裕靖, 伊藤 真利子, 青柳 岳司, 大西 立顕
<Corresponding author E-Mill:> hironobukawaguchi(at)rikkyo.ac.jp
<Abstract:> The simulation to obtain stress-strain curves of block copolymers requires large computational resources. As an alternative to simulation, a method for high-throughput prediction of stress-strain curves using a three-dimensional convolutional neural network has been reported. In this study, we incorporated shortcut coupling into the neural network and performed pre-training and transfer learning in a step-by-step manner to successfully predict the stress-strain curve with high accuracy while reducing the training cost.
<Keywords:> Block copolymers, Microphase separation, Stress-strain curve, Deep learning, 3D convolutional neural network
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0037/_article/-char/ja/