[Published online Journal of Computer Chemistry, Japan Vol.19, 94-98, by J-STAGE]
<Title:> 経路積分分子動力学法を用いたミューオニウム化分子の理論的解析
<Author(s):> 大場 優生, 河津 励, 立川 仁典
<Corresponding author E-Mill:> tachi(at)yokohama-cu.ac.jp
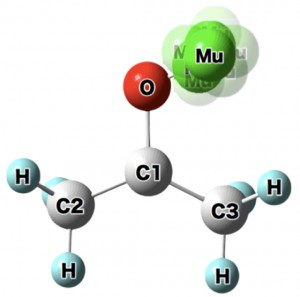
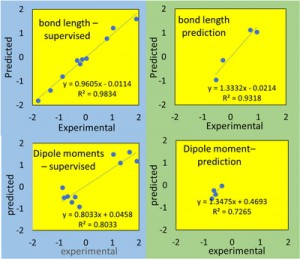
<Abstract:> 経路積分分子動力学(PIMD)計算により,ミューオニウム化アセトン分子(Mu-ACE)における超微細結合定数(HFCC)を算出した.アセトン酸素原子とミューオニウム間距離(ROMu)の伸長方向のポテンシャルの非調和性のもとで,ミューオンの大きな量子効果のためにROMuは伸長することが分かった.この際,ミューオンがラジカル電子を奪って中性Mu原子としての配置が出現することにより,HFCC値が増大することも分かった.水素化アセトン分子(H-ACE)に対しても同様な計算を実施しところ,HFCC値の実験値の大小関係を定性的に再現することができた.
<Keywords:> Keyword Muoniated Acetone Radicals, Hydrogenated Acetone Radicals, μSR, Hyperfine Coupling Constant (HFCC), Path Integral Molecular Dynamics, Nuclear Quantum Effect (NQE)
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0017/_article/-char/ja/
<Title:> 経路積分分子動力学法を用いたミューオニウム化分子の理論的解析
<Author(s):> 大場 優生, 河津 励, 立川 仁典
<Corresponding author E-Mill:> tachi(at)yokohama-cu.ac.jp
<Abstract:> 経路積分分子動力学(PIMD)計算により,ミューオニウム化アセトン分子(Mu-ACE)における超微細結合定数(HFCC)を算出した.アセトン酸素原子とミューオニウム間距離(ROMu)の伸長方向のポテンシャルの非調和性のもとで,ミューオンの大きな量子効果のためにROMuは伸長することが分かった.この際,ミューオンがラジカル電子を奪って中性Mu原子としての配置が出現することにより,HFCC値が増大することも分かった.水素化アセトン分子(H-ACE)に対しても同様な計算を実施しところ,HFCC値の実験値の大小関係を定性的に再現することができた.
<Keywords:> Keyword Muoniated Acetone Radicals, Hydrogenated Acetone Radicals, μSR, Hyperfine Coupling Constant (HFCC), Path Integral Molecular Dynamics, Nuclear Quantum Effect (NQE)
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/3/19_2020-0017/_article/-char/ja/