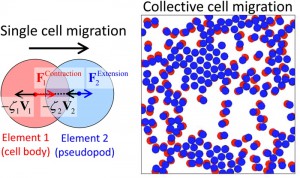
基板上で遊走 増殖する細胞集団のモデリング [Published online J. Comput. Chem. Jpn., 17, 14-19, by J-STAGE]
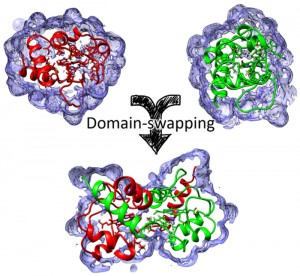
シトクロムcの多量体形成に関する理論的研究 [Published online J. Comput. Chem. Jpn., 17, 8-13, by J-STAGE]
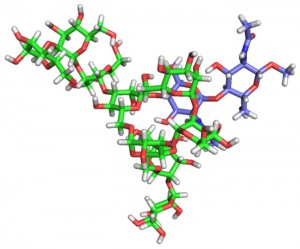
分子動力学計算とNMR計測を用いた糖鎖の配座空間探査 [Published online J. Comput. Chem. Jpn., 17, 1-7, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.17, 1-7, by J-STAGE]
<Title:> 分子動力学計算とNMR計測を用いた糖鎖の配座空間探査
<Author(s):> 山口 拓実, 渡邉 東紀男, 矢木 宏和, 加藤 晃一
<Corresponding author E-Mill:> takumi(at)jaist.ac.jp
<Abstract:> 糖鎖は複雑な分岐構造と高い内部運動の自由度をもち,その立体構造は水中で絶えず揺動している.したがって,糖鎖の生物機能発現に関する分子科学的基盤を 正しく理解するためには,立体構造を動態として描象することが重要である.核磁気共鳴(NMR)法と分子シミュレーションを組み合わせた動的構造 解析法の確立により,糖鎖のコンフォメーションをその揺らぎを含めて定量的に理解することが可能となってきた.常磁性NMR法とレプリカ交換分子 動力学計算法を用いて,タンパク質の品質管理に関わる一連のオリゴ糖鎖のコンフォメーションを明らかにした.さらに,配座空間探査に基づき,糖鎖 とそれを認識するタンパク質との結合様式を解析することで,静的な構造解析だけでは捉えることができない動的な相互作用機構の理解を進めることが できた.
<Keywords:> Oligosaccharide, NMR, Paramagnetic effect, Replica-exchange molecular dynamics simulation, Conformation analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/1/17_2018-0011/_html/-char/ja/
<Title:> 分子動力学計算とNMR計測を用いた糖鎖の配座空間探査
<Author(s):> 山口 拓実, 渡邉 東紀男, 矢木 宏和, 加藤 晃一
<Corresponding author E-Mill:> takumi(at)jaist.ac.jp
<Abstract:> 糖鎖は複雑な分岐構造と高い内部運動の自由度をもち,その立体構造は水中で絶えず揺動している.したがって,糖鎖の生物機能発現に関する分子科学的基盤を 正しく理解するためには,立体構造を動態として描象することが重要である.核磁気共鳴(NMR)法と分子シミュレーションを組み合わせた動的構造 解析法の確立により,糖鎖のコンフォメーションをその揺らぎを含めて定量的に理解することが可能となってきた.常磁性NMR法とレプリカ交換分子 動力学計算法を用いて,タンパク質の品質管理に関わる一連のオリゴ糖鎖のコンフォメーションを明らかにした.さらに,配座空間探査に基づき,糖鎖 とそれを認識するタンパク質との結合様式を解析することで,静的な構造解析だけでは捉えることができない動的な相互作用機構の理解を進めることが できた.
<Keywords:> Oligosaccharide, NMR, Paramagnetic effect, Replica-exchange molecular dynamics simulation, Conformation analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/17/1/17_2018-0011/_html/-char/ja/
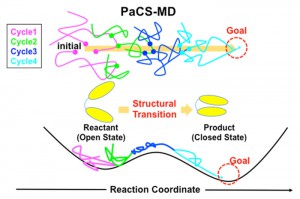
カスケード選択型分子動力学法によるタンパク質機能の動的秩序解析 [Published online in advanced , by J-STAGE]
DNAの電子状態計算 [Published online J. Comput. Chem. Jpn., 16, 157-159, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 157-159, by J-STAGE]
<Title:> DNAの電子状態計算
<Author(s):> 寺前 裕之, 青木 百合子
<Corresponding author E-Mill:> teramae(at)gmail.com
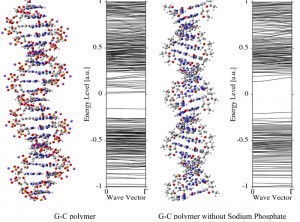
<Abstract:> As an electronic structure calculation of the B-type model-DNA, the calculation of (poly-(guanine) poly-(cytosine)) model polymer is performed by means of the ab initio crystal orbital method adapting the screw axis-symmetry which results in great reduction of computational efforts. All sugar backbones and sodium phosphate are included in the calculations. Energy band structures are calculated at the 6-31G level. For a comparison, the calculation without sodium phosphate is also performed. The resultant energy band structure is very different from that of the original one and it should be concluded that the alkali phosphate is necessary to describe the electronic structure of model-DNA.
<Keywords:> Crystal orbital method, Screw-axis symmetry, B-type DNA, Parallel processing, Energy band structure
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0052/_html/-char/ja/
<Title:> DNAの電子状態計算
<Author(s):> 寺前 裕之, 青木 百合子
<Corresponding author E-Mill:> teramae(at)gmail.com
<Abstract:> As an electronic structure calculation of the B-type model-DNA, the calculation of (poly-(guanine) poly-(cytosine)) model polymer is performed by means of the ab initio crystal orbital method adapting the screw axis-symmetry which results in great reduction of computational efforts. All sugar backbones and sodium phosphate are included in the calculations. Energy band structures are calculated at the 6-31G level. For a comparison, the calculation without sodium phosphate is also performed. The resultant energy band structure is very different from that of the original one and it should be concluded that the alkali phosphate is necessary to describe the electronic structure of model-DNA.
<Keywords:> Crystal orbital method, Screw-axis symmetry, B-type DNA, Parallel processing, Energy band structure
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0052/_html/-char/ja/
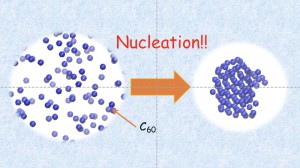
A Molecular Dynamics Study of the Temperature Effects on the Gas-Phase Crystal Growth of C60 Fullerene Molecules [Published online J. Comput. Chem. Jpn., 16, 152-154, by J-STAGE]
植物由来の香料化合物の分類に関する電子状態インフォマティクス研究 [Published online J. Comput. Chem. Jpn., 16, 155-156, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 155-156, by J-STAGE]
<Title:> 植物由来の香料化合物の分類に関する電子状態インフォマティクス研究
<Author(s):> 杉本 学, 空閑 瞳
<Corresponding author E-Mill:> sugimoto(at)kumamoto-u.ac.jp
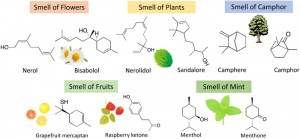
<Abstract:> We carried out electronic structure calculations on molecules having scent, and evaluated their electronic descriptors. Based on the calculated descriptors, a correlation between electronic similarities and scents of the molecules was investigated. Through this analysis, we found that a good correlation exists between these characteristics.
<Keywords:> キーワード:電子状態インフォマティクス, 電子的記述子, 電子的類似性, 香り, 香料
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0066/_html/-char/ja/
<Title:> 植物由来の香料化合物の分類に関する電子状態インフォマティクス研究
<Author(s):> 杉本 学, 空閑 瞳
<Corresponding author E-Mill:> sugimoto(at)kumamoto-u.ac.jp
<Abstract:> We carried out electronic structure calculations on molecules having scent, and evaluated their electronic descriptors. Based on the calculated descriptors, a correlation between electronic similarities and scents of the molecules was investigated. Through this analysis, we found that a good correlation exists between these characteristics.
<Keywords:> キーワード:電子状態インフォマティクス, 電子的記述子, 電子的類似性, 香り, 香料
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0066/_html/-char/ja/
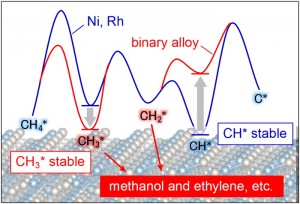
メタン活性化を目指したインフォマティクス [Published online J. Comput. Chem. Jpn., 16, 147-148, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 147-148, by J-STAGE]
<Title:> メタン活性化を目指したインフォマティクス
<Author(s):> 蒲池 高志, 斎藤 雅史, 辻 雄太, 吉澤 一成
<Corresponding author E-Mill:> kamachi(at)fit.ac.jp
<Abstract:> We investigated the C H bond cleavage of methane on various binary alloys using periodic density functional theory (DFT) calculations for catalyst screening. Cohesive energy, which strongly correlates with activation energy and heat of reaction for the C H bond cleavage, was computed for 337 alloys in AFLOW database to enable rapid screening.
<Keywords:> メタン転換触媒, 密度汎関数法, 触媒インフォマティクス
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0058/_html/-char/ja/
<Title:> メタン活性化を目指したインフォマティクス
<Author(s):> 蒲池 高志, 斎藤 雅史, 辻 雄太, 吉澤 一成
<Corresponding author E-Mill:> kamachi(at)fit.ac.jp
<Abstract:> We investigated the C H bond cleavage of methane on various binary alloys using periodic density functional theory (DFT) calculations for catalyst screening. Cohesive energy, which strongly correlates with activation energy and heat of reaction for the C H bond cleavage, was computed for 337 alloys in AFLOW database to enable rapid screening.
<Keywords:> メタン転換触媒, 密度汎関数法, 触媒インフォマティクス
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0058/_html/-char/ja/
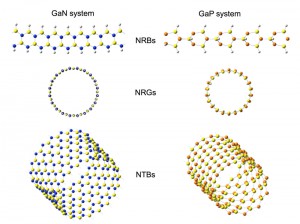
III-V族ヘテロ6員環からなるGaPおよびGaNナノ構造体の分子構造と電子構造 [Published online J. Comput. Chem. Jpn., 16, 149-151, by J-STAGE]
[Published online Journal of Computer Chemistry, Japan Vol.16, 149-151, by J-STAGE]
<Title:> III-V族ヘテロ6員環からなるGaPおよびGaNナノ構造体の分子構造と電子構造
<Author(s):> 小路 謙介, 松永 雄樹, 武田 京三郎
<Corresponding author E-Mill:> takeda(at)waseda.jp
<Abstract:> We computationally design GaN and GaP heteroatom nanostructures of nanoribbons (NRBs), nanorings (NRGs), and nanotubes (NTBs), and study the atomistic and electronic structures theoretically. First-principles calculations demonstrate that GaN finite NRBs have a flat molecular plane whereas GaP NRBs break the flatness of the NRB molecular planes. Although an NRG is produced by rolling an NRB (head to tail), the GaP system produces a specific NRG having a “magic ring numbe” whereas the GaN system can freely change the NRG diameter. A NTB stacked by these NRGs has a potential to be a one-dimensional semiconductor having a band gap of 1.5 3 eV and effective mass ratios 0.3 1.7 eV for an electron and a hole.
<Keywords:> GaN, GaP, NRB, NRG, NTB, Electronic and Molecular Structures, First-principles calculation
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0060/_html/-char/ja/
<Title:> III-V族ヘテロ6員環からなるGaPおよびGaNナノ構造体の分子構造と電子構造
<Author(s):> 小路 謙介, 松永 雄樹, 武田 京三郎
<Corresponding author E-Mill:> takeda(at)waseda.jp
<Abstract:> We computationally design GaN and GaP heteroatom nanostructures of nanoribbons (NRBs), nanorings (NRGs), and nanotubes (NTBs), and study the atomistic and electronic structures theoretically. First-principles calculations demonstrate that GaN finite NRBs have a flat molecular plane whereas GaP NRBs break the flatness of the NRB molecular planes. Although an NRG is produced by rolling an NRB (head to tail), the GaP system produces a specific NRG having a “magic ring numbe” whereas the GaN system can freely change the NRG diameter. A NTB stacked by these NRGs has a potential to be a one-dimensional semiconductor having a band gap of 1.5 3 eV and effective mass ratios 0.3 1.7 eV for an electron and a hole.
<Keywords:> GaN, GaP, NRB, NRG, NTB, Electronic and Molecular Structures, First-principles calculation
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/5/16_2017-0060/_html/-char/ja/