[Published online Journal of Computer Chemistry, Japan Vol.16, 63-69, by J-STAGE]
<Title:> 反復構造を持つ高分子に対する分子動力学計算のためのパラメータ設定支援プログラムo2pの開発
<Author(s):> 矢部 誠, 園部 智彩, 上田 一義, 武田 穣
<Corresponding author E-Mill:> takeda-minoru-bd(at)ynu.ac.jp
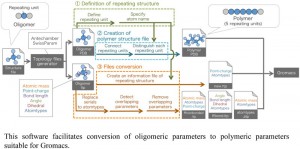
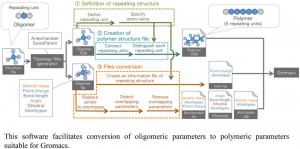
<Abstract:> Molecular dynamics (MD)の実行に必須の結合角や電荷などのパラメータセット(力場)がライブラリに存在しない場合,力場の量子力学計算による算出やパラメータ自動生成 ツールの活用という手法が用いられる.しかし,これらの手法は原子数の多い高分子の場合には適していない.そこで,3つの単位構造(両末端の単位 と中央部の繰り返し単位)を含むオリゴマーの力場を算出し,それらをポリマー形式(両末端部+中央部 × n)に変換し対処することがよく行われる.しかしながら,変換は手作業にならざるを得ず,非効率的でヒューマンエラーの恐れもある.そこで,反復構造を持 つ高分子に対するMDを簡便かつ確実に実行すべく,分子動力学ソフトとして汎用されているGromacs用の力場変換半自動化プログラムo2pを 開発した.開発にあたっては,作業負荷の低減と時間短縮を意図してGUIを積極的に取り入れた.また,複雑なファイル変換過程を自動化することに よってヒューマンエラーの一掃を目指した.本プログラムにより変換したファイルを用いてノナン中のアミロース分子を想定した系でMDを実行したと ころ,実験による報告と同じく左巻きの一重らせん構造が形成され,プログラムが有効に動作していることを確認した.
<Keywords:> Molecular dynamics simulation, Macromolecules, Parameters, Gromacs, Polymer, Repeating unit
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/3/16_2017-0013/_article/-char/ja/
<Title:> 反復構造を持つ高分子に対する分子動力学計算のためのパラメータ設定支援プログラムo2pの開発
<Author(s):> 矢部 誠, 園部 智彩, 上田 一義, 武田 穣
<Corresponding author E-Mill:> takeda-minoru-bd(at)ynu.ac.jp
<Abstract:> Molecular dynamics (MD)の実行に必須の結合角や電荷などのパラメータセット(力場)がライブラリに存在しない場合,力場の量子力学計算による算出やパラメータ自動生成 ツールの活用という手法が用いられる.しかし,これらの手法は原子数の多い高分子の場合には適していない.そこで,3つの単位構造(両末端の単位 と中央部の繰り返し単位)を含むオリゴマーの力場を算出し,それらをポリマー形式(両末端部+中央部 × n)に変換し対処することがよく行われる.しかしながら,変換は手作業にならざるを得ず,非効率的でヒューマンエラーの恐れもある.そこで,反復構造を持 つ高分子に対するMDを簡便かつ確実に実行すべく,分子動力学ソフトとして汎用されているGromacs用の力場変換半自動化プログラムo2pを 開発した.開発にあたっては,作業負荷の低減と時間短縮を意図してGUIを積極的に取り入れた.また,複雑なファイル変換過程を自動化することに よってヒューマンエラーの一掃を目指した.本プログラムにより変換したファイルを用いてノナン中のアミロース分子を想定した系でMDを実行したと ころ,実験による報告と同じく左巻きの一重らせん構造が形成され,プログラムが有効に動作していることを確認した.
<Keywords:> Molecular dynamics simulation, Macromolecules, Parameters, Gromacs, Polymer, Repeating unit
<URL:> https://www.jstage.jst.go.jp/article/jccj/16/3/16_2017-0013/_article/-char/ja/