[Published online Journal of Computer Chemistry, Japan -International Edition Vol.10, -, by J-STAGE]
<Title:> A Study of Deep Learning for Quantitative Analysis of Vitamin A in Cattle Blood
<Author(s):> Mizuki SHIBASAKI, Tetsuhito SUZUKI, Moriyuki FUKUSHIMA, Shin-ichi NAGAOKA, Yuichi OGAWA, Naoshi KONDO
<Corresponding author E-Mill:> nagaoka.shinichi.3c(at)kyoto-u.ac.jp
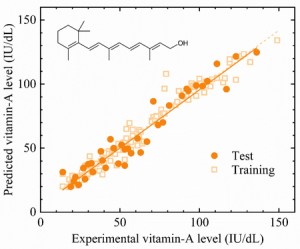
<Abstract:> Deep learning in combination with fluorescence excitation-emission spectroscopy was studied to quantitatively analyze vitamin A (retinol) in cattle blood. The neural network model being obtained with the deep learning predicted the vitamin-A levels with a coefficient of determination (R2) of 0.93 with respect to the experimental values. The combination of the deep learning and fluorescence excitation-emission spectroscopy has a potential to predict the vitamin-A level in the cattle blood accurately, rapidly and inexpensively and to improve production of marbled beef with maintaining cattle health. It could also be applied to quantitative vitamin-A assays of various biological tissues, foods and so on as well as to those of blood samples besides cattle.
<Keywords:> Deep learning, Neural network, Fluorescence excitation-emission spectroscopy, Vitamin A, Retinol, Cattle blood, random forest
<URL:> https://www.jstage.jst.go.jp/article/jccjie/10/0/10_2024-0012/_html
<Title:> A Study of Deep Learning for Quantitative Analysis of Vitamin A in Cattle Blood
<Author(s):> Mizuki SHIBASAKI, Tetsuhito SUZUKI, Moriyuki FUKUSHIMA, Shin-ichi NAGAOKA, Yuichi OGAWA, Naoshi KONDO
<Corresponding author E-Mill:> nagaoka.shinichi.3c(at)kyoto-u.ac.jp
<Abstract:> Deep learning in combination with fluorescence excitation-emission spectroscopy was studied to quantitatively analyze vitamin A (retinol) in cattle blood. The neural network model being obtained with the deep learning predicted the vitamin-A levels with a coefficient of determination (R2) of 0.93 with respect to the experimental values. The combination of the deep learning and fluorescence excitation-emission spectroscopy has a potential to predict the vitamin-A level in the cattle blood accurately, rapidly and inexpensively and to improve production of marbled beef with maintaining cattle health. It could also be applied to quantitative vitamin-A assays of various biological tissues, foods and so on as well as to those of blood samples besides cattle.
<Keywords:> Deep learning, Neural network, Fluorescence excitation-emission spectroscopy, Vitamin A, Retinol, Cattle blood, random forest
<URL:> https://www.jstage.jst.go.jp/article/jccjie/10/0/10_2024-0012/_html