[Advanced Published online Journal of Computer Chemistry, Japan, by J-STAGE]
<Title:> 固体表面に存在する水素原子が示す量子効果
<Author(s):> 奥山 弘
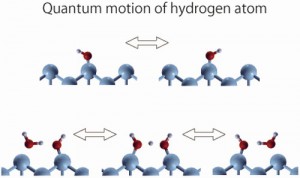
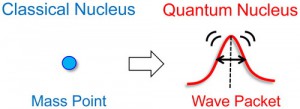
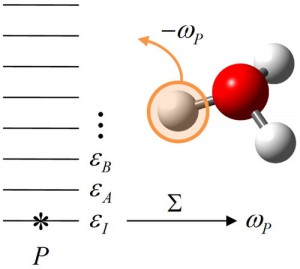
<Abstract:> Quantum effect is manifest for hydrogen atoms on surfaces as well as for those in solution and solids. The excited states of a hydrogen atom directly adsorbed on metal surfaces are beyond the diffusion barrier, thus being delocalized to the next sites, and even spread over the entire surface in the low-coverage limit. In addition, water and hydroxyl group on the surfaces show significant quantum effect, due to tunneling and zero-point motion of the hydrogen atom. We briefly review some experimental and theoretical studies that revealed the quantum effect of the hydrogen atom on the surfaces.
<Keywords:> Hydrogen atom, Metal surface, Quantum delocalization
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2016-0013/_article/-char/ja/
<Title:> 固体表面に存在する水素原子が示す量子効果
<Author(s):> 奥山 弘
<Abstract:> Quantum effect is manifest for hydrogen atoms on surfaces as well as for those in solution and solids. The excited states of a hydrogen atom directly adsorbed on metal surfaces are beyond the diffusion barrier, thus being delocalized to the next sites, and even spread over the entire surface in the low-coverage limit. In addition, water and hydroxyl group on the surfaces show significant quantum effect, due to tunneling and zero-point motion of the hydrogen atom. We briefly review some experimental and theoretical studies that revealed the quantum effect of the hydrogen atom on the surfaces.
<Keywords:> Hydrogen atom, Metal surface, Quantum delocalization
<URL:> https://www.jstage.jst.go.jp/article/jccj/advpub/0/advpub_2016-0013/_article/-char/ja/