[Published online Journal of Computer Chemistry, Japan -International Edition Vol.3, -, by J-STAGE]
<Title:> Thermodynamics of Three-phase Equilibrium by Van Der Waals Equation of State
<Author(s):> Yosuke KATAOKA
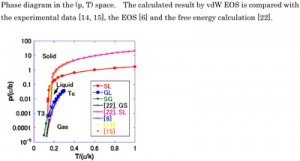
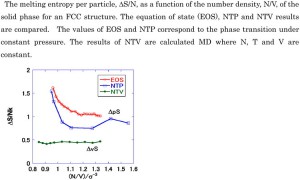
<Abstract:> The phase diagram for a three-phase equilibrium was derived using van der Waals (vdW) equation of state (EOS) with respect to pressure. Aside from the typical liquid-gas vdW EOS, a new solid-gas vdW EOS was introduced. The new vdW EOS had the same functional form as the original equation of state and only the van der Waals coefficients were different. The thermodynamic EOSs were integrated to obtain the internal energy and the integral constants were given explicitly. The calculated phase diagram was consistent with that for argon and the Lennard-Jones system.
<Keywords:> Three-phase equilibrium, Van der Waals equation of state, Argon, Lennard-Jones system, Thermodynamics
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0026/_html
<Title:> Thermodynamics of Three-phase Equilibrium by Van Der Waals Equation of State
<Author(s):> Yosuke KATAOKA
<Abstract:> The phase diagram for a three-phase equilibrium was derived using van der Waals (vdW) equation of state (EOS) with respect to pressure. Aside from the typical liquid-gas vdW EOS, a new solid-gas vdW EOS was introduced. The new vdW EOS had the same functional form as the original equation of state and only the van der Waals coefficients were different. The thermodynamic EOSs were integrated to obtain the internal energy and the integral constants were given explicitly. The calculated phase diagram was consistent with that for argon and the Lennard-Jones system.
<Keywords:> Three-phase equilibrium, Van der Waals equation of state, Argon, Lennard-Jones system, Thermodynamics
<URL:> https://www.jstage.jst.go.jp/article/jccjie/3/0/3_2017-0026/_html