[Published online Journal of Computer Chemistry, Japan Vol.18, 18-28, by J-STAGE]
<Title:> リチウムイオン電池Solid Electrolyte Interphase (SEI)に関する第一原理計算研究
<Author(s):> 館山 佳尚
<Corresponding author E-Mill:> TATEYAMA.Yoshitaka(at)nims.go.jp
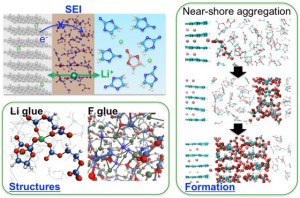
<Abstract:> リチウムイオン電池(LIB)中の負極と電解液の界面に主に充電時に形成される被膜をSolid Electrolyte Interphase (SEI)と呼ぶ.このSEIの性質によってLIBの性能や安全性が大きく変化する.しかし,この界面過程のその場観察・オペランド観察実験はいまだに困難が多く,高精度な理論計算による微視的機構の解明に大きな期待が寄せられている.本稿では,特に密度汎関数理論をベースにした第一原理計算によるSEI研究の現状を,著者らの研究も含めて,概観し今後について展望する.
<Keywords:> Keywords Lithium ion battery, Solid electrolyte Interphase, first-principles calculation, redox reactions, interface
<URL:> https://www.jstage.jst.go.jp/article/jccj/18/1/18_2018-0046/_article/-char/ja/