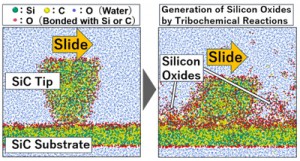
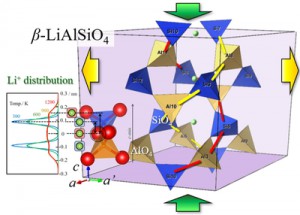
[Published online Journal of Computer Chemistry, Japan Vol.19, 164-166, by J-STAGE]
<Title:> 人工知能を用いた打音試験による片状及び球状黒鉛鋳鉄の材質判定
<Author(s):> 篠原 美月, 内田 希, 岩見 祐貴, 平本 雄一, 加藤 雅也, 菅野 利猛
<Corresponding author E-Mill:> mitsuki_shinohara(at)gmail.com
<Abstract:> Tapping test is an inspection method that determines the presence or absence of abnormality in a specimen based on the difference in the sound when tapping a material. This method is used to inspect buildings and railway vehicles. It is considered that this method can be used for quality evaluation of cast iron. However, although the tapping test has the advantage of being able to be performed non-destructively and simply, it also has the disadvantage of requiring a worker who can distinguish sounds. In order to solve this problem, we introduced a neural network and studied whether it is possible to judge the quality of cast iron by learning the tapping sound of cast iron.
<Keywords:> Flake graphite cast iron, Spheroidal graphite cast iron, Neural network, Tapping test
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0016/_article/-char/ja/
<Title:> 人工知能を用いた打音試験による片状及び球状黒鉛鋳鉄の材質判定
<Author(s):> 篠原 美月, 内田 希, 岩見 祐貴, 平本 雄一, 加藤 雅也, 菅野 利猛
<Corresponding author E-Mill:> mitsuki_shinohara(at)gmail.com
<Abstract:> Tapping test is an inspection method that determines the presence or absence of abnormality in a specimen based on the difference in the sound when tapping a material. This method is used to inspect buildings and railway vehicles. It is considered that this method can be used for quality evaluation of cast iron. However, although the tapping test has the advantage of being able to be performed non-destructively and simply, it also has the disadvantage of requiring a worker who can distinguish sounds. In order to solve this problem, we introduced a neural network and studied whether it is possible to judge the quality of cast iron by learning the tapping sound of cast iron.
<Keywords:> Flake graphite cast iron, Spheroidal graphite cast iron, Neural network, Tapping test
<URL:> https://www.jstage.jst.go.jp/article/jccj/19/4/19_2021-0016/_article/-char/ja/