[Published online Journal of Computer Chemistry, Japan Vol.14, 184-185, by J-STAGE]
<Title:> 生分解性キレート剤が作る鉄(III)錯体の構造予測
<Author(s):> 大友 秀一, 崎山 博史
<Corresponding author E-Mill:> saki(at)sci.kj.yamagata-u.ac.jp
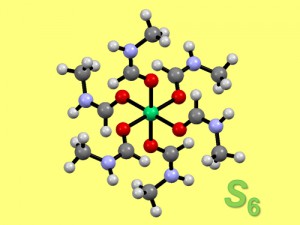
<Abstract:> The structures of iron(III) complexes with biodegradative chelating agent, methylglycinediacetic acid [H3(mgda)], were computationally predicted based on the DFT method. The structures of dinuclear iron(III) complexes, [Fe2O(mgda)2(CO3)]4 and [Fe2O(mgda)2(H2O)4]2 , were predicted, and the structures were thought to be similar to those of related dinuclear iron(III) complexes with nitrilotriacetate (nta3 ), [Fe2O(nta)2(CO3)]4 and [Fe2O(nta)2(H2O)4]2 , respectively. The mgda complex, [Fe2O(mgda)2(H2O)4]2 , is thought to interact with hydrogen peroxide like the nta complex [Fe2O(nta)2(H2O)4]2 .
<Keywords:> 生分解性キレート剤, 鉄(III)錯体, 構造予測, 異性体, 密度汎関数法
<URL:> https://www.jstage.jst.go.jp/article/jccj/14/6/14_2015-0070/_article/-char/ja/
<Title:> 生分解性キレート剤が作る鉄(III)錯体の構造予測
<Author(s):> 大友 秀一, 崎山 博史
<Corresponding author E-Mill:> saki(at)sci.kj.yamagata-u.ac.jp
<Abstract:> The structures of iron(III) complexes with biodegradative chelating agent, methylglycinediacetic acid [H3(mgda)], were computationally predicted based on the DFT method. The structures of dinuclear iron(III) complexes, [Fe2O(mgda)2(CO3)]4 and [Fe2O(mgda)2(H2O)4]2 , were predicted, and the structures were thought to be similar to those of related dinuclear iron(III) complexes with nitrilotriacetate (nta3 ), [Fe2O(nta)2(CO3)]4 and [Fe2O(nta)2(H2O)4]2 , respectively. The mgda complex, [Fe2O(mgda)2(H2O)4]2 , is thought to interact with hydrogen peroxide like the nta complex [Fe2O(nta)2(H2O)4]2 .
<Keywords:> 生分解性キレート剤, 鉄(III)錯体, 構造予測, 異性体, 密度汎関数法
<URL:> https://www.jstage.jst.go.jp/article/jccj/14/6/14_2015-0070/_article/-char/ja/