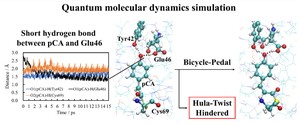
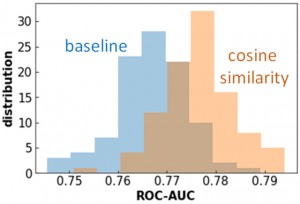
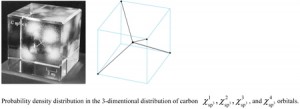
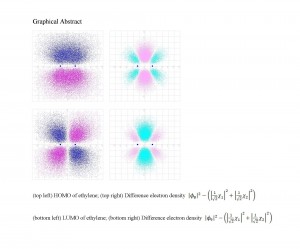
[Published online Journal of Computer Chemistry, Japan Vol.22, 28-30, by J-STAGE]
<Title:> 量子多体系ダイナミクスシミュレータの構築
<Author(s):> 戸畑 海研, 石田 邦夫
<Corresponding author E-Mill:> ishd_kn(at)cc.utsunomiya-u.ac.jp
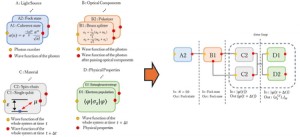
<Abstract:> We propose a quantum dynamics simulator for light-irradiated matter. As the coherent control of the quantum states of matter is an intriguing problem in realizing quantum technology, the quantum properties of light-matter complexes have attracted much attention. Theoretical calculations corresponding to individual experimental configurations are required to understand these properties, and simulation methods that flexibly adapt to various experimental configurations are required. In this paper, we propose a quantum dynamics simulation method that encompasses program modules corresponding to light sources, optical components, material systems, and the evaluation of physical properties. We demonstrate the entanglement generation dynamics between two spin-chains via irradiated photons and show that a flexible and expandable quantum dynamics simulator can be constructed.
<Keywords:> Quantum many-body system simulator, Quantum dynamics calculation, Modularized simulator
<URL:> https://www.jstage.jst.go.jp/article/jccj/22/2/22_2023-0024/_article/-char/ja/
<Title:> 量子多体系ダイナミクスシミュレータの構築
<Author(s):> 戸畑 海研, 石田 邦夫
<Corresponding author E-Mill:> ishd_kn(at)cc.utsunomiya-u.ac.jp
<Abstract:> We propose a quantum dynamics simulator for light-irradiated matter. As the coherent control of the quantum states of matter is an intriguing problem in realizing quantum technology, the quantum properties of light-matter complexes have attracted much attention. Theoretical calculations corresponding to individual experimental configurations are required to understand these properties, and simulation methods that flexibly adapt to various experimental configurations are required. In this paper, we propose a quantum dynamics simulation method that encompasses program modules corresponding to light sources, optical components, material systems, and the evaluation of physical properties. We demonstrate the entanglement generation dynamics between two spin-chains via irradiated photons and show that a flexible and expandable quantum dynamics simulator can be constructed.
<Keywords:> Quantum many-body system simulator, Quantum dynamics calculation, Modularized simulator
<URL:> https://www.jstage.jst.go.jp/article/jccj/22/2/22_2023-0024/_article/-char/ja/