[Published online Journal of Computer Chemistry, Japan Vol.20, 94-96, by J-STAGE]
<Title:> HSP90とコシャペロンp23複合体の安定性に関するシミュレーション研究
<Author(s):> 松倉 里紗, 宮下 尚之
<Corresponding author E-Mill:> miya(at)waka.kindai.ac.jp
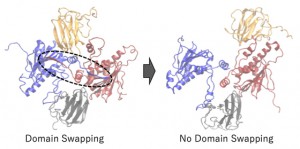
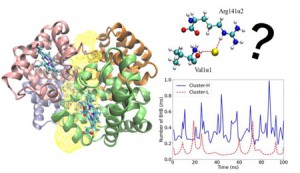
<Abstract:> Heat Shock Protein 90 (HSP90) has been known as one of the molecular chaperone proteins, and it supports the correct folding of client proteins. It has been getting attention as the target protein for an anti-cancer drug because the client proteins sometimes include the critical proteins of progressing cancer. In the HSP90 chaperon cycle, several co-chaperones and substrates interact with HSP90 in turn. In this study, we focused on the interaction between HSP90 and p23, which is one of the co-chaperones. After the HSP90 dimer forms the closed conformation, the p23 and ATP bind to the HSP90. However, the detailed interaction among them has not been clear yet. To clear the mechanism of the interaction among HSP90, ATP, and p23, we performed the all-atom molecular dynamics simulations of ATP bound/unbound HSP90 with/without p23. We found that the domain swapping in HSP90 closed conformation is essential for the stable binding between HSP90 and p23, and the ATP binding supports the stability of the HSP90 and p23 complex.
<Keywords:> Molecular Dynamics Simulation, Co-chaperone, Heat Shock Protein 90, Chaperone cycle, Computational Biophysics
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0044/_article/-char/ja/
<Title:> HSP90とコシャペロンp23複合体の安定性に関するシミュレーション研究
<Author(s):> 松倉 里紗, 宮下 尚之
<Corresponding author E-Mill:> miya(at)waka.kindai.ac.jp
<Abstract:> Heat Shock Protein 90 (HSP90) has been known as one of the molecular chaperone proteins, and it supports the correct folding of client proteins. It has been getting attention as the target protein for an anti-cancer drug because the client proteins sometimes include the critical proteins of progressing cancer. In the HSP90 chaperon cycle, several co-chaperones and substrates interact with HSP90 in turn. In this study, we focused on the interaction between HSP90 and p23, which is one of the co-chaperones. After the HSP90 dimer forms the closed conformation, the p23 and ATP bind to the HSP90. However, the detailed interaction among them has not been clear yet. To clear the mechanism of the interaction among HSP90, ATP, and p23, we performed the all-atom molecular dynamics simulations of ATP bound/unbound HSP90 with/without p23. We found that the domain swapping in HSP90 closed conformation is essential for the stable binding between HSP90 and p23, and the ATP binding supports the stability of the HSP90 and p23 complex.
<Keywords:> Molecular Dynamics Simulation, Co-chaperone, Heat Shock Protein 90, Chaperone cycle, Computational Biophysics
<URL:> https://www.jstage.jst.go.jp/article/jccj/20/3/20_2021-0044/_article/-char/ja/