[Published online Journal of Computer Chemistry, Japan Vol.21, 33-35, by J-STAGE]
<Title:> β-LiAlSiO4結晶の分子動力学法に用いる原子間相互作用の改良
<Author(s):> 大垣 毅弥, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)mmm.muroran-it.ac.jp
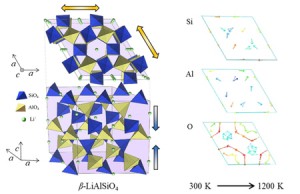
<Abstract:> Thermal change of lattice parameters of β-LiAlSiO4 crystal simulated by molecular dynamics simulation was improved by revision of the interatomic potential. The discontinuity of thermal change of c-axis lattice parameter observed in the previous work between 800 K and 900 K was dissolved, but the simulated linear thermal expansion of c-axis was smaller than the reference data. The visualized shift of relative coordinates of each atom with the temperature increase from 300 K to 1200 K showed the different variation between the two types of double helix structures that exist in the unit cell.
<Keywords:> Molecular dynamics, ??i??β??/i??-eucryptite, ??i??β??/i??-LiAlSiO4, Thermal expansion, Structure analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/2/21_2022-0023/_article/-char/ja/
<Title:> β-LiAlSiO4結晶の分子動力学法に用いる原子間相互作用の改良
<Author(s):> 大垣 毅弥, 澤口 直哉
<Corresponding author E-Mill:> nasawa(at)mmm.muroran-it.ac.jp
<Abstract:> Thermal change of lattice parameters of β-LiAlSiO4 crystal simulated by molecular dynamics simulation was improved by revision of the interatomic potential. The discontinuity of thermal change of c-axis lattice parameter observed in the previous work between 800 K and 900 K was dissolved, but the simulated linear thermal expansion of c-axis was smaller than the reference data. The visualized shift of relative coordinates of each atom with the temperature increase from 300 K to 1200 K showed the different variation between the two types of double helix structures that exist in the unit cell.
<Keywords:> Molecular dynamics, ??i??β??/i??-eucryptite, ??i??β??/i??-LiAlSiO4, Thermal expansion, Structure analysis
<URL:> https://www.jstage.jst.go.jp/article/jccj/21/2/21_2022-0023/_article/-char/ja/