[Published online Journal of Computer Chemistry, Japan Vol.25, 1-6, by J-STAGE]
<Title:> Anisotropic Displacement Parameters of Ba2 in Clathrate Structure Ba8Ga16Ge30 from Molecular Dynamics Simulations
<Author(s):> Yoyo HINUMA
<Corresponding author E-Mill:> y.hinuma(at)aist.go.jp
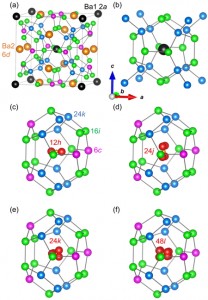
<Abstract:> Atomic displacement parameters (ADPs) are crystallographic structural data that may represent atomic motion, possible static displacive disorder, and thermal vibration. Thermoelectric materials often contain rattling atoms to improve performance, and ADPs describe how atoms rattle. This paper discusses the ADPs of the Ba2 site of Ba8Ga16Ge30, a clathrate structure thermoelectric material. A molecular dynamics simulation approach using a neural network potential was applied to Ba8Ga16Ge30 models considering the disorder of Ga and Ge on the Ga/Ge cage sites. The calculated U is non-zero when extrapolated to 0 K, overestimates experimental U11 by 0.005-0.009 2 at temperatures between 200 and 300 K, and overestimates experimental U22 by 0.010-0.014 2 between 15 and 300 K.
<Keywords:> Atomic displacement parameters, clathrate structure, Ba8Ga16Ge30, molecular dynamics, universal neural network potential
<URL:> https://www.jstage.jst.go.jp/article/jccj/25/1/25_2025-0026/_article/-char/ja/
<Title:> Anisotropic Displacement Parameters of Ba2 in Clathrate Structure Ba8Ga16Ge30 from Molecular Dynamics Simulations
<Author(s):> Yoyo HINUMA
<Corresponding author E-Mill:> y.hinuma(at)aist.go.jp
<Abstract:> Atomic displacement parameters (ADPs) are crystallographic structural data that may represent atomic motion, possible static displacive disorder, and thermal vibration. Thermoelectric materials often contain rattling atoms to improve performance, and ADPs describe how atoms rattle. This paper discusses the ADPs of the Ba2 site of Ba8Ga16Ge30, a clathrate structure thermoelectric material. A molecular dynamics simulation approach using a neural network potential was applied to Ba8Ga16Ge30 models considering the disorder of Ga and Ge on the Ga/Ge cage sites. The calculated U is non-zero when extrapolated to 0 K, overestimates experimental U11 by 0.005-0.009 2 at temperatures between 200 and 300 K, and overestimates experimental U22 by 0.010-0.014 2 between 15 and 300 K.
<Keywords:> Atomic displacement parameters, clathrate structure, Ba8Ga16Ge30, molecular dynamics, universal neural network potential
<URL:> https://www.jstage.jst.go.jp/article/jccj/25/1/25_2025-0026/_article/-char/ja/